 2023, Vol. 34
2023, Vol. 34 


b College of Chemistry and Chemical Engineering, Jishou University, Jishou 416000, China
 Xin Wang was born in Heilongjiang, China and received her MS degree from Northeast Normal University in 2012. In July 2013, she joined the College of Chemistry and Chemical Engineering, Anyang Normal University. In 2019, she pursued her Ph.D. degree in Zhengzhou University. Her research program is drug design, structural identification and structural modification of natural products; Xin Wang was born in Heilongjiang, China and received her MS degree from Northeast Normal University in 2012. In July 2013, she joined the College of Chemistry and Chemical Engineering, Anyang Normal University. In 2019, she pursued her Ph.D. degree in Zhengzhou University. Her research program is drug design, structural identification and structural modification of natural products; |
 Jianping Meng was born in Heilongjiang Province, China in 1994. In 2014, she began her studies at the School of Science of Heihe University. In 2019, she pursued her MS degree under the supervision of Kai Sun in YanTai University. Her current research is focused on green synthetic chemistry; Jianping Meng was born in Heilongjiang Province, China in 1994. In 2014, she began her studies at the School of Science of Heihe University. In 2019, she pursued her MS degree under the supervision of Kai Sun in YanTai University. Her current research is focused on green synthetic chemistry; |
 Kai Sun was born in Shanxi, China in 1983. He received his Ph.D. degree in organic chemistry from Northeast Normal University in 2013 under the supervision of Prof. Qian Zhang. In July 2020, he joined the College of Chemistry and Chemical Engineering, YanTai University, where he is an associate professor. His current research is focused on radical C–H functionalization and green synthetic chemistry Kai Sun was born in Shanxi, China in 1983. He received his Ph.D. degree in organic chemistry from Northeast Normal University in 2013 under the supervision of Prof. Qian Zhang. In July 2020, he joined the College of Chemistry and Chemical Engineering, YanTai University, where he is an associate professor. His current research is focused on radical C–H functionalization and green synthetic chemistry |
Organic sulfur and selenium compounds are omnipresent in a large number of natural products, pharmaceutical molecules, and chiral ligands, and they have received considerable attention over the past few decades on account of their wide range of biological and medicinal properties [1-11]. Sulfones have gradually become an indispensable part of modern organic chemistry and have been widely used in the transformation and synthesis of various drugs because of their special electronic and unique structural properties [12]. Particularly, chalcogenative sulfones are important organic compounds possessing both sulfonyl (R1SO2-) and sulfur (R2S-)/seleno (R3Se-) groups [13-16]. Four selenosulfonates synthesized from o-nitrobenzeneselenyl bromide and sodium arenesulfinates were first proposed by Foss's group in 1947 [17]. Although thiosulfonates and selenosulfonates were first reported 70 years ago, research on their applications was not actively conducted at that time [18-27]. Thiosulfonates generally exhibit low toxicity and have served as efficient reactants for the synthesis of many valuable sulfur-containing compounds over the last few years [5,28-30]. In addition, thiosulfonates and selenosulfonates can be used as practical sulfonating reagents in many chemical conditions [18,21,24]. For these reasons, thiosulfonates and selenosulfonates have become very valuable synthons. In recent years, considerable efforts have been made by chemists to develop new and efficient methods for synthesizing thiosulfonates and selenosulfonates, as well as applying them to new reactions to construct novel sulfur- or selenium-containing compounds.
At present, thiosulfonates and selenosulfonates are involved in a wide range of reactions, including electrophilic reactions, nucleophilic reactions, and radical reactions. Among them, the rapid development of free-radical chemistry is a hot spot in chemical synthesis that cannot be underestimated [31-43]. As new, effective, and stable free-radical reagents, chalcogenative sulfones are involved in some radical reactions that are worthy of special attention. Although there are several reviews on the applications of thiosulfonates and selenosulfonates, these related methodologies mainly highlight the synthesis, reactivity, and activation modes of fluoroalkyl thiosulfonates and selenosulfonates [44], or S-trifluoroethyl benzenesulfonothioates as bench-stable reagents for electrophilic trifluoroethylthiolation [45]. In 2020, Maes's group contributed a timely and elegant paper on the synthesis and applications of thiosulfonates, in which nucleophilic, electrophilic, radical, coupling, polymerization, and multicomponent reactions were all briefly mentioned [46]. Herein, we mainly focus on and summarize the synthesis and applications strategies of thiosulfonates and selenosulfonates as stable radical reagents, which has not been reviewed so far in a general way. In this paper, the synthetic methods of chalcogenative sulfones are briefly summarized, and the research progress of radical reactions involving thiosulfonates and selenosulfonates is reviewed comprehensively. In addition, in order to further understand the recent research progress, reaction scopes, applications, limitations, and some mechanisms are also briefly discussed.
2. Synthesis of chalcogenative sulfonesAs highlighted in the Introduction, thiosulfonates and selenosulfonates have interesting properties and are also widely used as versatile sulfenylating reagents in many organic transformations. For these reasons, chemists have made considerable efforts to design and develop new and effective methods for the construction of thiosulfonates and selenosulfonates. However, literature investigation indicates that the synthetic methods of thiosulfonates and selenosulfonates are limited. We briefly summarize some of the known synthetic strategies based on the precursors used to generate thiosulfonates and selenosulfonates. Each of these reactions will be discussed in summary in the following introduction.
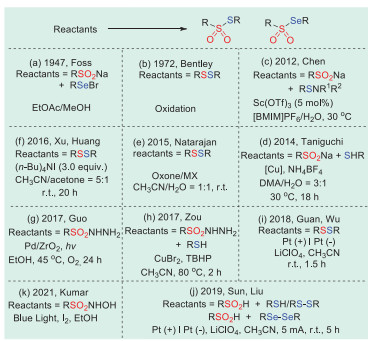
In 1947, Foss's group first discovered the synthesis of selenosulfonates from o-nitrobenzeneselenylbromide and sodium arenesulfinates (Scheme 1a) [17]. Unfortunately, only four examples of selenosulfonates have been successfully obtained in this way.

|
Download:
|
| Scheme 1. Different synthetic methods of thiosulfonates and selenosulfonates. | |
{kind=link}
In 1972, Bentley and colleagues converted sodium sulfinates and alkyl disulfides directly into thiosulfonates using stoichiometric amounts of silver nitrate (Scheme 1b) [47]. However, the substrate range of this method was limited and only suitable for sodium methanesulfinates.
In 2012, Chen's group proposed the first example of sodium sulfinates with N-(organothio)succinimides catalyzed by Sc(OTf)3 under ionic liquids and water cosolvent conditions, affording thiosulfonates with considerate yields (Scheme 1c) [48].
In 2014, Taniguchi et al. developed a method for the preparation of thiosulfonates catalyzed by copper using sodium sulfinates in an oxygen atmosphere (Scheme 1d) [49].
One year later, Natarajan's group introduced a new method in which oxone combines with MX (MX = KBr, KCl, NaBr, or NaCl) to oxidize aliphatic or aromatic disulfides containing electron-donating or electron-withdrawing groups into corresponding thiosulfonates (Scheme 1e) [50]. Compared with other synthetic methods, the outstanding advantages of this method include mild conditions, short reaction times, and avoiding the use of toxic and unstable reagents.
A practical synthesis of electrophilic thiosulfonates and disulfides in the presence of tetrabutylammonium iodide by selecting suitable solvents using cheap and readily available sulfonyl chlorides as starting materials was developed by Xu's and Huang's groups in 2016 (Scheme 1f) [51]. The method had high efficiency, economy, and mild conditions, which was attractive for the preparation of these two sulfur-containing compounds.
In 2017, Guo et al. used a Pd/ZrO2 nanocomposite photocatalyst to catalyze the decomposition of sulfonyl hydrazines and synthesized thiosulfonates under mild conditions without oxidants or promoters (Scheme 1g) [52]. In the same year, Zou's group described a convenient tert-butyl hydroperoxide (TBHP)-mediated radical cross coupling of sulfonylhydrazides with thiols catalyzed by CuBr2 to produce thiosulfonates with moderate to excellent yields (Scheme 1h) [53]. The method had a great tolerance for different functional groups and provided many kinds of thiosulfonates.
In 2018, Guan, Wu et al. proposed a novel, efficient, and environmentally friendly strategy for the preparation of thiosulfonates by the electrochemical oxidation of thiols (Scheme 1i) [54]. This method enabled the formation of thiosulfonates without catalysts and oxidants. The reaction conditions were simple and mild, and could be used at gram scale; they were also suitable for the late-stage synthesis of bioactive molecules.
In 2019, Sun, Liu et al. realized the electrochemical oxidative cross dehydrogenation coupling of arylsulfonic acids with thiophenol compounds by a radical method, providing a series of unsymmetrical thiosulfonates with excellent yields (Scheme 1j) [55]. At the same time, the electrochemical method could also be applied to the reaction of arylsulfinic acids and disulfides or diselenides to access thiosulfonates or selenosulfonates, respectively.
In 2021, Kumar and co-workers established a strategy for the successful synthesis of a series of symmetrical benzene thiosulfonates by irradiation of N-hydroxy sulphonamides under visible light induction in the presence of ethanol solvent (Scheme 1k) [56]. This method used ubiquitous blue light as the reaction condition, which was a relatively mild and environmentally friendly method.
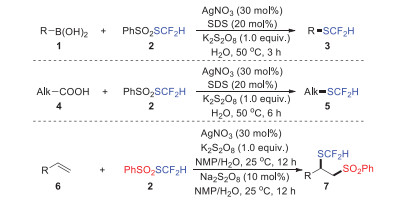
3. Applications of thiosulfonates and selenosulfonates 3.1. Applications of thiosulfonates and selenosulfonates based on metal catalysisThe transition metal-catalyzed radical reaction strategy of chalcogenative sulfones had become an indispensable tool for the construction of heteroatom–S(Se) bonds due to its rapid and efficient characteristics, especially in the construction of structurally diverse compounds [57-65]. In 2016, Shen and Lu's group discovered a stable radical reagent S-(difluoromethyl)benzenesulfonothioate (PhSO2SCF2H), which could realize the radical difluoromethylthiolation of aryl and alkyl boronic acids 1, the decarboxylative difluoromethylthiolation of aliphatic acids 4, and the difunctionalization of alkenes 6 under mild conditions (Scheme 2) [66]. The strategy was characterized by simple operation, mild conditions, and the ability to access the target products in moderate to excellent yields. Due to the mild reaction conditions, various common functional groups such as fluorines, chlorines, bromines, amides, and nitros were compatible. The functionalization strategy involving PhSO2SCF2H as a free-radical agent opened up a new direction for the construction of difluoromethyl compounds.

|
Download:
|
| Scheme 2. Construction of difluoromethyl compounds catalyzed by copper. | |
{kind=link}
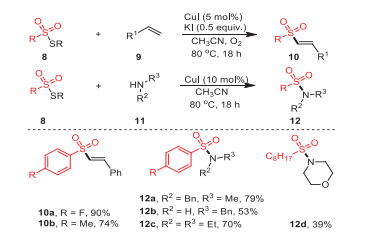
In 2017, Jang's group established a strategy of directly coupling thiosulfonates with alkenes and amines, respectively, breaking the original S–S bond and forming new C(sp2)–S/N–S bonds, thus constructing a series of synthetically and pharmaceutically useful vinyl sulfones 10 and sulfonamides 12 with synthetic and medicinal value (Scheme 3) [67]. Notably, thiosulfonate derivatives containing both electron-donating and electron-withdrawing groups offered the desired products with moderate to excellent yields. Surprisingly, octyl-substituted thiosulfonate yielded the desired product 12d with 39% yield, where it was speculated that aliphatic thiosulfonate might not be suitable for this transformation.

|
Download:
|
| Scheme 3. The synthesis of vinyl sulfones and sulfonamides catalyzed by copper catalyst. | |
{kind=link}
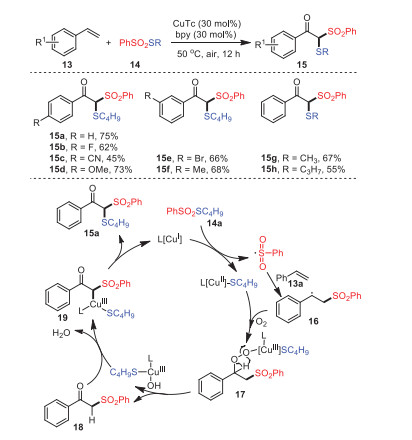
A method to construct β-ketone thiosulfones 15 by oxidative trifunctionalization of olefins 13 was proposed by Xu's group in 2018 (Scheme 4) [68]. The conversion proceeded through molecular oxygen activation under the catalytic action of copper, forming two new C–S bonds in a single operation under mild conditions. Under standard conditions, various electronically rich, neutral, and poor styrenes were all well tolerated, and the corresponding β-keto thioether products were obtained in moderate yields.

|
Download:
|
| Scheme 4. The formation of functionalized β-keto thiosulfones. | |
{kind=link}
Based on the results of some control experiments and literature precedents, a plausible reaction mechanism was proposed, as shown in Scheme 3. S-Butyl benzenesulfonothioate 14a was catalyzed by copper(Ⅰ) to form the sulfonyl radical, and then reacted with styrene to generate radical intermediate 16. In the presence of L-[Cu]SC4H9, intermediate 16 was transformed into radical 17 through activating molecular oxygen. The fragmentation of radical 17 offered β-keto sulfone 18, which reacted with the removed L-[CuOH]+ to form intermediate 19, and subsequent reductive elimination revealed the cause of the formation of the final product 15a.
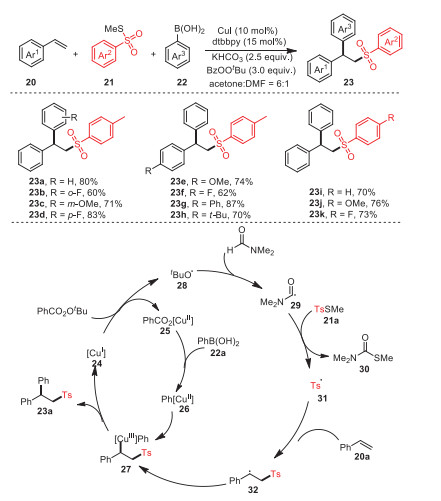
In 2020, Jia et al. developed an effective strategy for the preparation of a wide range of 2, 2-diarylethyl sulfone derivatives 23 by the Cu-catalyzed intermolecular difunctionalization of styrenes 20 with methyl thiosulfonates 21 and arylboronic acids 22 (Scheme 5) [69]. Remarkably, the mild nature of the approach made it suitable for late-stage modification of biologically active natural products, and the different substrate scopes demonstrated the functional group's tolerance for this reaction. In addition, a series of relevant control experiments were carried out to verify the mechanism of this reaction. The formation of the product was significantly inhibited by the addition of the radical scavenger 2, 2, 6, 6-tetramethylpiperidinyl 1-oxide (TEMPO) under standard conditions, demonstrating that the transformation involved a radical pathway.

|
Download:
|
| Scheme 5. The preparation of 2, 2-diarylethyl sulfones. | |
{kind=link}
At first, Cu(Ⅰ) 24 reacted with BzOOtBu to form copper complex 25 and radical 28, which then extracted hydrogen from DMF to access formyl radical 29. Afterwards, radical 31 obtained via a radical relay process was added to styrene 20a to produce benzyl radical 32. Cu(Ⅲ) intermediate 27 was obtained by oxidative trapping of radical 32 and aryl copper(Ⅱ) species 26 formed by the transmetalation of Cu(Ⅱ) complex 25 with arylboronic acid 22a. In the end, reductive elimination of the Cu(Ⅲ) intermediate 27 accounted for the formation of final product 23a.
3.2. Applications of chalcogenative sulfones under visible light irradiationOver the past decade, many chemists have been devoted to researching atom transfer radical addition (ATRA) reactions catalyzed by visible light, as photoredox catalysis was characterized as a clean, efficient, and environmentally friendly method, and as an effective tool under mild conditions [70-76]. In recent years, visible light-induced chemical transformations had received increasing attention due to their important role in synthetic chemistry. Due to the special reaction mode of visible-light photoredox catalysis, many transformations that were previously unattainable by transition metal catalytic processes became possible. Next, the applications of visible light-induced chalcogenative sulfones as free-radical reagents are introduced in detail. This series of methods has also been successfully used in the construction of various chain or cyclic compounds, which fully reflects the bright prospects of photochemistry.
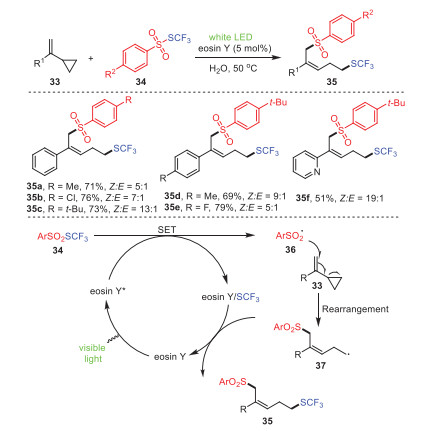
In 2020, Xu et al. reported a metal-free organocatalytic system mediated by visible light to achieve the remote difunctionalization of vinylcyclopropane 33 involving group migration with ArSO2SCF3 reagents 34, accessing the expected difunctional products 35 with moderate yields (Scheme 6) [77]. This transformation demonstrated a good tolerance towards functional groups, as well as excellent regioselectivity and stereoselectivity. Obviously, substrates with electron-withdrawing groups at the para position of their phenyl rings had slightly higher yields than electron-donating groups. Fortunately, the stereoselectivity of the target product was significantly improved to 13:1 when S-(trifluoromethyl)-4-(tert-butyl)benzenesulfonate 35c was used as a difunctionalization reagent. This pathway enabled the 1, 5-trifluomethylthio-sulfonylation of vinyl-cyclopropane using eosin Y as a catalyst, which is cheaper and more tolerated than transition-metal or redox-catalyst systems, and water as a "green" solvent. Considering the mild conditions of this reaction, its atomic efficiency, its environmentally friendly solvent, and the recyclability of the catalytic system, it is speculated that this protocol has strong application prospects in terms of green indicators. Mechanistically, eosin Y went into its excited state eosin Y* by irradiation with visible light; then, substrate 34 was excited by it to generate radical 36, and the eosin Y/SCF3 complex was simultaneously formed. Further reaction of radical 36 with substrate 33 was followed by a rearrangement to form intermediate 37, which was then reacted with the eosin Y/SCF3 complex to obtain final product 35.

|
Download:
|
| Scheme 6. Organocatalytic 1, 5-trifluomethylthio-sulfonylation of vinylcyclopropane mediated by visible light in water phase. | |
{kind=link}
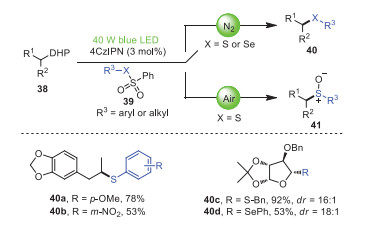
In the same year, Wang, Ji et al. realized a novel synthesis route for the construction of C(sp3)–S or C(sp3)–Se bonds by the visible-light-promoted cross-linking of 4-alkyl-1, 4-dihydropyridines 38 with thiosulfonates/selenosulfonates 39 in the absence of transition metals and additives, oxidants, or bases (Scheme 7) [78]. This strategy was characterized by readily available substrates, mild reaction conditions, and excellent chemoselectivity, which could also be well generalized to the synthesis of thiolated or selenylated glycosides that have not been explored before. Notably, both electron-donating (OMe) and electron-withdrawing (NO2) substituents were well tolerated in the para- and meta-positions of S-aryl benzenesulfonothioates, so that products 40a and 40b could be isolated in 78% and 53% yields, respectively. Surprisingly, S-benzylbenzenesulfonate and Se-aryl benzenesulfonate were also suitable for this visible-light-promoted reaction in moderate to excellent yields and selectivity.

|
Download:
|
| Scheme 7. Visible-light-promoted construction of thiolated and selenylated glycosides. | |
{kind=link}
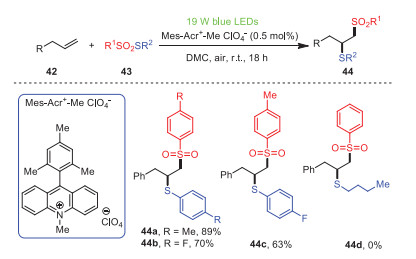
Soon after, Maes et al. developed a metal-free method of generating sulfonyl and sulfenyl radicals simultaneously through energy transfer of photo-organocatalysts excited by visible light followed by vicinal thiosulfonylation of unactivated olefins 42 under visible light irradiation (Scheme 8) [79]. This strategy provided a novel method for the synthesis of extensive 1, 2-thiosulfonylation products 44 by using energy transfer rather than single electron transfer. It is worth noting that this method did not contain metals and oxidants and could be carried out in green solvents in an air atmosphere, which was suitable for the functionalization of alkenes. Additionally, the substituents could be successfully tolerated, whether they were electron-donating groups or electron-withdrawing groups, and target products could be obtained with moderate to excellent yields. Unfortunately, S-butyl benzenethiosulfonate did not yield the expected product 44d under the same condition. This procedure can be carried out under mild conditions without additional metals and oxidants at room temperature, so it may be more attractive from an environmental and economical point of view.

|
Download:
|
| Scheme 8. Thiosulfonylation of unactivated alkenes with visible-light organic photocatalysis. | |
{kind=link}
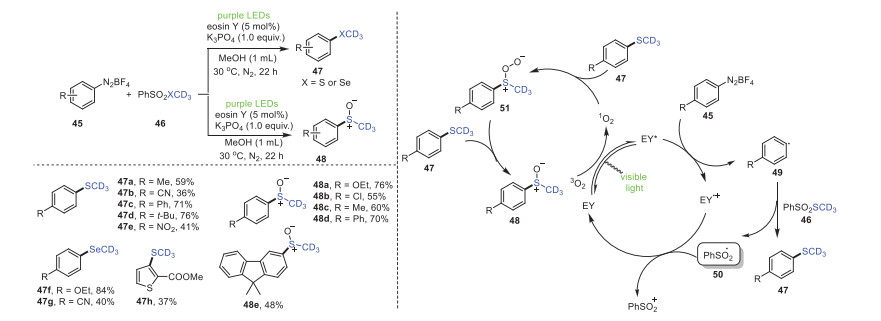
Later, a new, effective, and mild visible-light catalytic green method for aryldiazonium salts 45 via deuterated thiomethylation/methylselenation was revealed by Wang's group (Scheme 9) [80]. Remarkably, aryl diazo salt derivatives with functional groups, such as methyl 45a, cyano 45b, phenyl 45c, tert-butyl 45d and nitro 45e, had good tolerance during this transformation process, generating the expected products with 97% D incorporation. Heteroaryldiazonium salts could also be suitable for this process under the same conditions, yielding the target products.

|
Download:
|
| Scheme 9. Synthesis of trideuteromethylated sulfides, sulfoxides and selenides under visible light irradiation. | |
{kind=link}
According to previous literature reports, a feasible reaction mechanism is proposed in Scheme 9. Initially, the formation of EY* by visible light irradiation reacts with aromatic diazonium salt 45 to furnish aryl radical 49 through a single electron transfer (SET) process. Subsequently, the sulfone radical 50 obtained by the reaction of the aryl radical 49 with PhSO2SCD3 reacts with the radical cation of the photocatalyst to access the sulfonyl cation and reproduce the photocatalyst (EY). Finally, 1O2, successfully obtained by energy transfer between 3O2 and EY*, could oxidize trideuteromethyl sulfide to obtain the final trideuteromethyl sulfoxide 48.
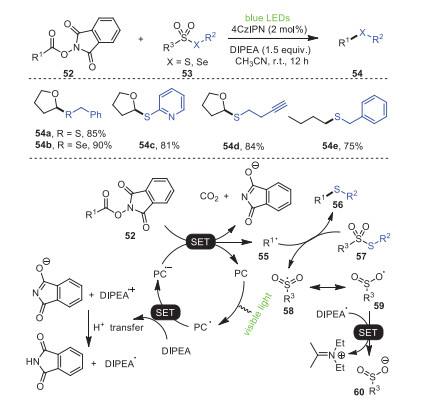
In 2020, Wang and coworkers disclosed a novel and efficient organophotoredox synthetic method, in which a C–S/Se bond was constructed by the reaction of thiosulfonates/selenosulfonates 53 with redox-active esters 52 (Scheme 10) [81]. Notably, a unique feature of this strategy was to use feedstock carboxylic-acid-derived RAEs 52 as radical progenitors, enabling an unprecedentedly wide range of substrates, which, in addition to its remarkable functional group tolerance, made this method a promising strategy for the construction of C–S bonds widely used in organic synthesis. The study of thiosulfonates as radical receptors of thioesters and RAEs as the universal radical precursors of C-glucosylation inspired this group to establish the visible light mediated thiolation of RAEs and thiosulfonates. One hypothesis was that the coupling of the radical 55 produced by the corresponding RAEs 52 with thiosulfonate 57 was the main reason for the synthesis of thioether.

|
Download:
|
| Scheme 10. Organophotoredox-catalyzed formation of C–S/Se bonds from coupling of redox-active esters with thiosulfonates/selenosulfonates. | |
{kind=link}
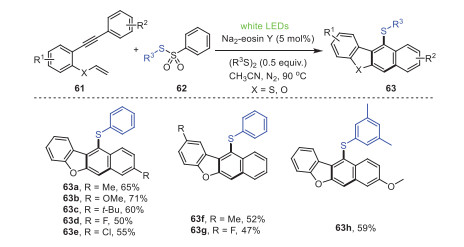
In 2021, the groups of Chen, Xu, and Mo described a visible light induced cascade reaction of 2-vinyloxy arylalkynes 61 and thiosulfonates 62 to construct a series of thio-substituted dibenzofuran derivatives 63 with certain yields (Scheme 11) [82]. This process provided a universal and mild route to construct a extensive range of thio-substituted dibenzofuran derivatives. Significantly, aryl alkyne substrates containing electron-donating substituents in the R1 position obtained higher yields than electron-withdrawing substituents. Mechanism studies showed that the thiosulfonylation product of 2-vinyloxy-arylalkyne was the key intermediate in this reaction, followed by cyclization/aromatization to produce the final product, in which the disulfide acted as a hydrogen extractor in the aromatization process.

|
Download:
|
| Scheme 11. Visible-light-induced catalysis for the synthesis of thio-substituted dibenzofuran derivatives. | |
{kind=link}
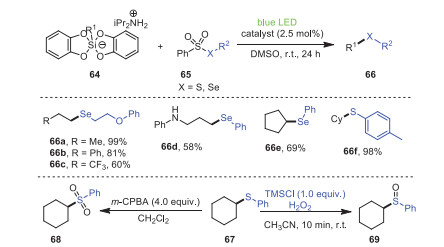
Almost 2 months later, Wang and coworkers successfully established a mild metal-free strategy in which a radical cross coupling of bis-catecholato silicon compounds 64 induced by visible light with seleno- or thiosulfonates 65 furnished a host of unsymmetric 1-alkyl-alkylselenide compounds 66 that were previously difficult to prepare (Scheme 12) [83]. This protocol could be used not only for the preparation of unsymmetric alkyl-alkylselenides, but also for the efficient construction of aryl-alkyl selenides, heteroayl-alkyl selenides, and aryl-alkyl sulfides. The catalytic system was compatible with electron-deficient and electron-rich substrates, or aliphatic and aromatic substrates, and the corresponding products could be successfully obtained in moderate to excellent yields. In addition, the yields of sulfoxide 69 and sulfoxide 68 of compound 67 treated with different oxidants were 88% and 78%, respectively, indicating that this protocol had great advantages in potential industrial applications.

|
Download:
|
| Scheme 12. Visible-light-promoted for the construction of unsymmetric 1-alkyl-alkylselenide compounds. | |
{kind=link}
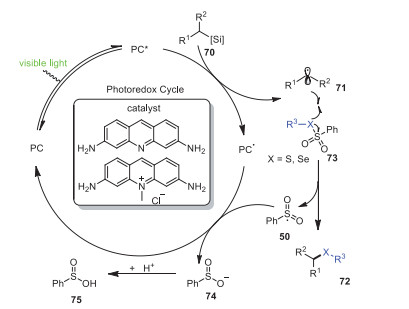
According to the experimental results above and previous literature reviews, a reasonable reaction mechanism is proposed in Scheme 13. Alkylsilicate 70 underwent single electron transfer to furnish alkyl radical 71 under the action of photocatalyst, and then reacted with selenosulfonate or sulfonothioate to obtain final product 72 and sulfone radical intermediate 50, respectively. Finally, benzosulfinic acid 75 was obtained by protonation of sulfone anionic intermediate 74 generated from reduction of the sulfone radical 50.

|
Download:
|
| Scheme 13. Proposed mechanism of the construction of unsymmetric 1-alkyl-alkylselenide compounds. | |
{kind=link}
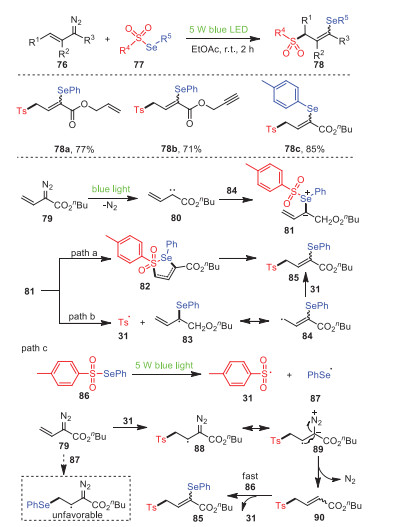
A visible-light-driven radical 1, 3-selenosulfonylation of vinyldiazo compounds 76 with selenosulfonates 77 without any photocatalysts or additives was achieved by Zhou's group in 2021, resulting in a wide variety of γ-seleno allylic sulfones 78 with high yields (Scheme 14) [84]. Obviously, allyl 78a and proparynyl 78b could be applied for this transformation. Furthermore, controlled experimental studies confirmed that this process was carried out through a radical chain propagation process, and the applications of the product were demonstrated by deselenization, reduction, bromination and allylation. On the basis of controlled and competitive experiments, a reasonable mechanism was depicted, as shown in Scheme 14.

|
Download:
|
| Scheme 14. Visible-light-driven catalysis for the synthesis of γ-seleno allylic sulfones. | |
{kind=link}
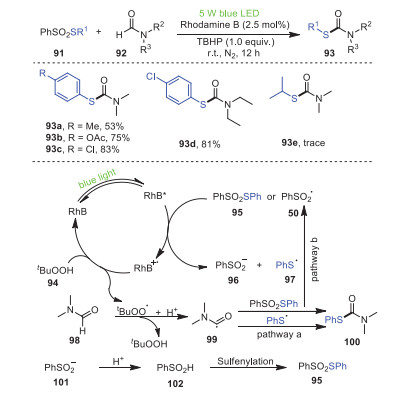
In 2021, Bi's, Feng's and Geng's groups developed a strategy to synthesize new C–S bonds promoted by visible light, using thiosulfonates 91 and N-substituted formamides 92 as starting materials, Rhodamine B as a photocatalyst, and tert-butylhydrogen peroxide (TBHP) as an oxidant to obtain secondary and tertiary thiocarbamates 93 under blue-light irradiation at room temperature (Scheme 15) [85]. In particular, this method used N-monosubstituted formamides as amide sources to synthesize secondary thiocarbonates, as a first example. In addition, S-arylbenzene thiosulfonates bearing electron-withdrawing groups had higher yields than those bearing electron-donating groups, most likely due to the strong electron-donating groups inhibiting the reaction process. Unfortunately, S-alkyl benzenethiosulfonate 93e was not suitable for this reaction under the optimized conditions, which was most likely caused by the instability of the sulfenyl radical.

|
Download:
|
| Scheme 15. Visible-light-promoted for the synthesis of secondary and tertiary thiocarbamates. | |
{kind=link}
Firstly, RhB* and 95 underwent an SET process to form radical cations RhB•+, benzenesulfinic acid anion 96, and benzenesulfenyl radical 97. Afterwards, RhB•+ withdrew an electron from TBHP, resulting in the formation of RhB, the tert-butylperoxyl radical and H+. Ultimately, the final product was formed by the coupling of acyl radical 99 and 97, in which acyl radical 99 was obtained by the extraction of hydrogen from DMF 98 by the tert-butylperoxyl radical (pathway a). Conversely, radical 99 reacted with 95 to form PhSO2 radical 50 as well as 100 (pathway b).
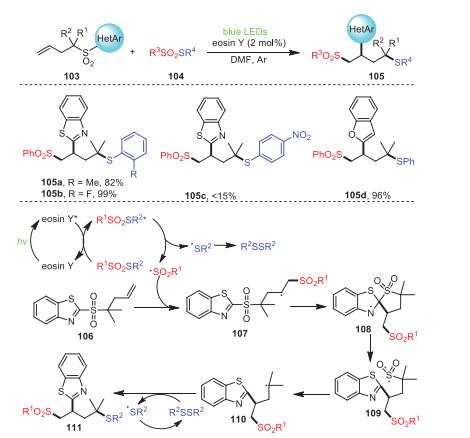
Soon later, a method for the formation of new C–S/Se bonds between butenyl benzothiazole sulfones 103 with thiosulfonates or selenosulfonates 104 via radical cascade sulfonylation/aryl migration/desulfonylation under visible light was established by Wang's group (Scheme 16) [86]. This process provided a new procedure for the acquisition of a series of 1, 2, 4-trifunctionalization products via a radical sulfonyl-smile rearrangement pathway. Notably, this strategy, using practical and economical eosin Y as the catalyst, could tolerate many functional groups, resulting in a series of 1, 2, 4-trifunctionalization products of butenyl benzothiazole sulfones. Surprisingly, S-(4-nitrophenyl) benzenesulfonothioate produced less than 15% of the expected product 105c due to its strong electron-withdrawing group.

|
Download:
|
| Scheme 16. Visible-light-triggered 1, 2, 4-trifunctionalization of butenyl benzothiazole sulfone with thiosulfonate/selenosulfonates. | |
{kind=link}
First of all, after irradiation, eosin Y was transformed into its excited state, which underwent energy transfer with thiosulfonate to generate its corresponding excited state, and the homolysis of the excited-state thiosulfonate produced a sulfonyl radical and a sulfenyl radical. Subsequently, the five-membered ring intermediate 108 was formed by radical addition of the terminal double bond of 106 and subsequent intramolecular radical cyclization. The re-aromatization of intermediate 108 and cleavage of the C–S bond explained the formation of intermediate 109. Eventually, 109 removed a molecule of SO2 to produce radical intermediate 110, which further reacted with disulfide to access the target product 111.
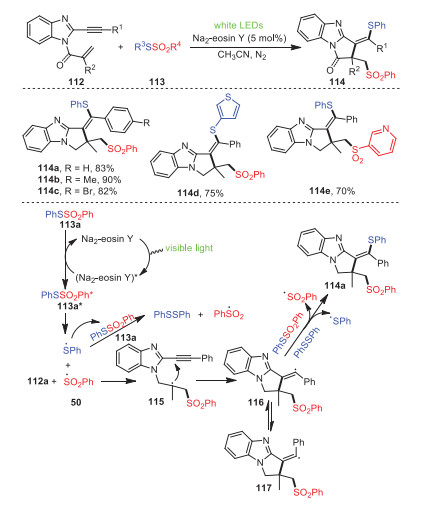
In 2021, Xu and Chen's group investigated a radical cascade reaction of 1-allyl-2-ethynylbenzoimidazoles 112 with thiosulfonates 113 induced by visible light, providing thiosulfonylated pyrro[1, 2-a]benzimidazoles 114 (Scheme 17) [87]. The transformation had the advantages of mild conditions, environmental friendliness, excellent functional group tolerance, and regioselectivity. The addition of radical inhibitor TEMPO under the standard reaction suggested that the transformation involved a radical process. Control experiments also showed that thiosulfonates were activated through a photocatalytic energy transfer pathway. Based on the results of relevant control experiments and previous references, a possible mechanism was depicted, as shown in Scheme 17.

|
Download:
|
| Scheme 17. Visible-light-induced construction of thiosulfonylated pyrro[1, 2-a]benzimidazoles. | |
{kind=link}
3.3. Applications of thiosulfonates and selenosulfonates by synergetic metal/photoredox catalysis
Combining two distinctive catalytic systems, photoredox catalysis mediated by visible light and transition-metal catalysis, makes it possible to achieve unprecedented reactions [88-101]. The combination of photoredox catalysis with transition metal catalysis, known as metallaphotoredox catalysis or synergetic metal/photoredox catalysis, has become a mainstay in synthetic methodology in the past decade [102]. In addition, the use of inexpensive transition metals as catalysts for photoreactions was an innovation and breakthrough for a series of previous noble-metal catalysis and visible-light catalysis experiments. This combination of metal catalysis and photooxidation-reduction strategies shed light on the further development of metalloradical reactions in organic synthesis.
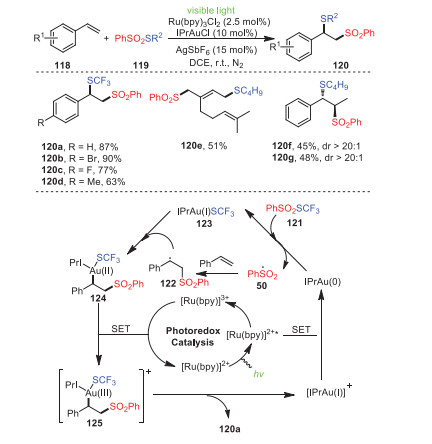
In 2017, Xu's group accomplished a unique gold and visible light photoredox synergistic catalysis difunctionalization of alkenes 118, achieving a series of difunctional products 120 with moderate to excellent yields (Scheme 18) [103]. Particularly, the synergistic combination of gold catalysis and photoredox catalysis was the key for the selective introduction of thio groups together with sulfonyl groups into olefins. Interestingly, the main products of the trans-120f and cis-olefins 120g had the same structure, as well as relatively similar yields with similar diastereoselectivity, which was an important advantage of this reaction.

|
Download:
|
| Scheme 18. Dual gold- and photoredox-catalyzed intermolecular atom transfer thiosulfonylation of alkenes. | |
{kind=link}
The proposed mechanism is depicted in Scheme 18. First, irradiation of Ru(bpy)32+ obtained its excited state Ru(bpy)32+*, which underwent an SET reaction with cationic IPrAu(Ⅰ) to generate Ru(bpy)33+ and IPrAu(0) and simultaneously initiate the catalytic cycle. Next, the highly active IPrAu(0) could reduce PhSO2SCF3 to IPrAu(Ⅰ)SCF3 and a benzenesulfonyl radical 50, which added to styrene, affording an alkyl radical 122. The alkyl radical could oxidize 123 to form intermediate 124, which was further oxidized by Ru(Bpy)33+ to an Au(Ⅲ) intermediate 125, producing Ru(bpy)32+, thus completing the photoredox catalytic cycle. Finally, reductive elimination of the Au(Ⅲ) intermediate 125 explained the formation of the desired product and led to the regeneration of the Au(Ⅰ) catalyst.
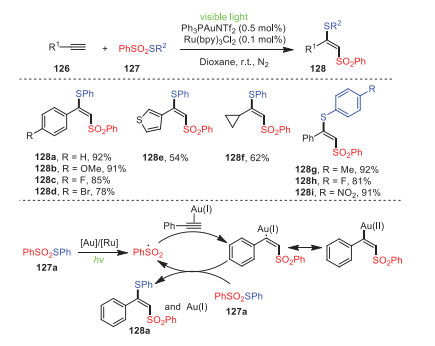
In 2019, Xu, Wei and coworkers established a strategy for the rapid construction of thio-functionalized vinylsulfones 128 by gold-photooxidation-reduction co-catalyzed thiosulfonylation of alkynes 126 (Scheme 19) [104]. Remarkably, various alkynes, such as aromatic alkynes and aliphatic alkynes, could react with thiosulfonates to produce the corresponding thio-functional products. Furthermore, aromatic alkynes possessing electron-donating groups at their para-position gave higher yields than those bearing electron-withdrawing groups. Mechanistically, under the co-catalysis of a gold catalyst and photocatalyst, PhSO2SPh underwent S–S bond cleavage to form a sulfonyl radical, which added to alkyne's triple bond to form a vinyl radical in the presence of the gold catalyst. Finally, radical propagation was carried out with 127a to obtain the target thiosulfonylation product and regenerate the Au(Ⅰ) catalyst.

|
Download:
|
| Scheme 19. Gold/photoredox-cocatalyzed atom transfer thiosulfonylation of alkynes. | |
{kind=link}
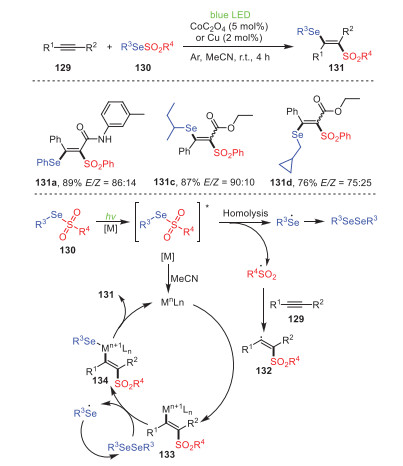
A visible-light-induced Co- or Cu-catalyzed 1, 2-selenosulfylation of alkynes 129 under mild conditions to construct a series of β-(seleno) vinyl sulfones 131 was proposed by Ji and Wang's group in the same year (Scheme 20) [105]. Notably, alkyamides possessing either electron-withdrawing groups or electron-donating groups reacted smoothly to produce a series of β-(seleno)vinyl sulfones with high yields. In addition, alkyl selenosulfonates (131b, 131c) were also suitable for this reaction, with corresponding yields of 87% and 76%, respectively. Most importantly, the protocol used inexpensive transition metals as catalysts to achieve the visible-light catalysis cycle, which was an innovation in a series of previous experiments on the co-catalysis of precious metals and visible light, but also a new economical and environmentally friendly catalytic pathway following visible-light catalysis.

|
Download:
|
| Scheme 20. Visible-light-induced Co- or Cu-catalyzed selenosulfonylation of alkynes. | |
{kind=link}
On the basis of controlled and competitive experiments, a reasonable mechanism was proposed, as shown in Scheme 20. Initially, in the catalysis of metal under visible light, 130 was transformed into its excited state, which underwent homolysis to form a sulfonyl radical and a seleno radical. Subsequently, the sulfonyl radical reacted with alkyne to access intermediate 132, which underwent coordination with the metal to furnish intermediate 133. Ultimately, reductive elimination of intermediate 134, formed by the interaction between intermediate 133 and R3SeSeR3, explained the formation of product 131 and regenerated the catalyst.
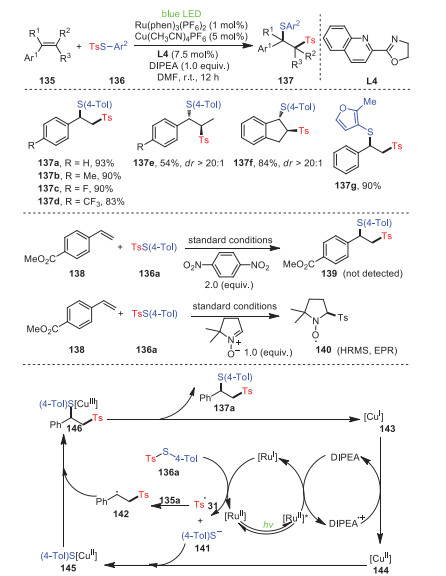
In 2021, Jia's and Liu's groups revealed the atom transfer radical addition of copper/photooxidation-reduction co-catalyzed styrenes 135 and thiosulfonates 136 (Scheme 21) [106]. Obviously, both the electron-withdrawing and electron-donating groups on styrenes exerted a negligible effect on the catalytic results. Furthermore, the styrene derivatives represented by β-methylstyrene 137e and cyclic indene 137f met the optimal catalytic conditions and had good regioselectivity and yields. In addition, the formation of 139 was completely inhibited by the addition of a radical scavenger (1, 4-dinitrobezene) from 138 and 136a under standard conditions. As expected, when 1.0 equiv. of 5, 5-dimethyl-1-pyrroline N-oxide was added to the same reaction system, the desired tosyl radical-trapped product 140 was found to be generated. The above mechanistic studies showed that the sulfonyl radical was the key intermediate of the transformation.

|
Download:
|
| Scheme 21. The construction of dibenzofulvene derivatives. | |
{kind=link}
3.4. Other applications of chalcogenative sulfones under metal-free conditions
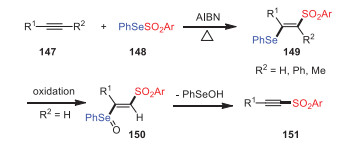
In this section, the reactions of selenosulfonates and thiosulfonates via breaking the original S(Se)-SO2R bond with the assistance of azo compounds, peroxides, heating, and organic small molecules are discussed. In 1983, Miura and Kobayashi observed that Se-phenyl p-tolueneselenosulfonates 148 underwent thermally induced 1, 2-additions to acetylenes, producing novel β-(phenylseleno)vinyl sulfones 149 in high yields (Scheme 22) [107]. The reaction had excellent regioselectivity and stereoselectivity, and it occurred through a radical chain mechanism initiated by the thermal decomposition of selenosulfonates.

|
Download:
|
| Scheme 22. The preparation of β-(phenylseleno)vinyl sulfones under metal-free conditions. | |
{kind=link}
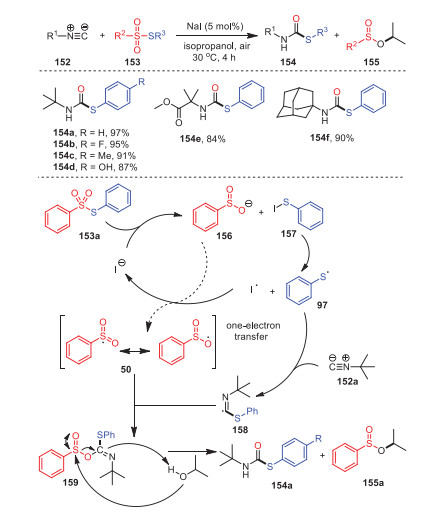
In 2016, Maes' and Ruijter's groups reported a new method for the construction of secondary thiocarbates 154 from isocyanides 152 and thiosulfonates 153 catalyzed by sodium iodide under mild conditions (Scheme 23) [108]. Remarkably, this reaction could be applied to substrates with electron-withdrawing or electron-donating substituents, resulting in a series of S-aryl tert-butylthiocarbamates with considerable yields. In addition to tert-butylisocyanide, other tertiary isocyanides could also participate in this transformation, and even adamantyl isocyanides 154f could be applied.

|
Download:
|
| Scheme 23. The formation of secondary thiocarbamates. | |
{kind=link}
Mechanistically, first, 153a reacted with sodium iodide to form benzenesulfinate anion 156 and PhSI 157, which underwent homolysis, forming radical 97. Subsequently, the intermediate 159 was successfully obtained through the reaction of radical 158 and 50 obtained by the addition of 97 and isocyanide 152a. Radical 158, derived from the addition of 97 with isocyanide 153a, reacted with intermediate 50 successfully to access intermediate 159. Ultimately, the alcoholysis of intermediate 159 with isopropanol accounted for the formation of 154a and 155a.
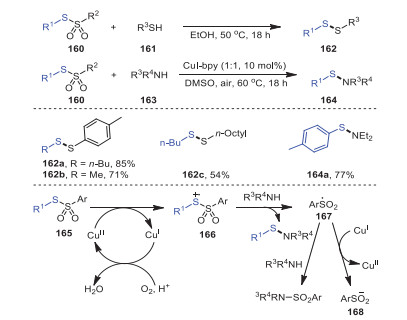
One year later, Taniguchi's group investigated a protocol for producing unsymmetrical disulfides 162 in high yields by reacting thiosulfonates 160 as free-radical reagents with nucleophile thiol reagents 161 under metal-free conditions (Scheme 24) [109]. Meanwhile, the reaction of amines 163 and thiosulfonates 160 to produce sulfanilamides 164 catalyzed by copper was also investigated. Notably, the conversion could obtain the desired unsymmetrical disulfides at excellent yields for both aryl-substituted and alkyl-substituted thiosulfonates. The addition of TEMPO, a radical scavenger, significantly inhibited the reaction process, suggesting that thiosulfonates reacted with amine through a radical reaction. Mechanistically, sulfonamide and a sulfonyl radical 167 were produced by the reaction of amine with radical intermediate 166 originating from the oxidation of thiosulfonate by a copper catalyst in air. The sulfonyl radical reacted with amine or was reduced by CuI to further yield the final product sulfonamide.

|
Download:
|
| Scheme 24. The construction of unsymmetrical disulfide and sulfenamide under metal-free conditions or promoted by copper. | |
{kind=link}
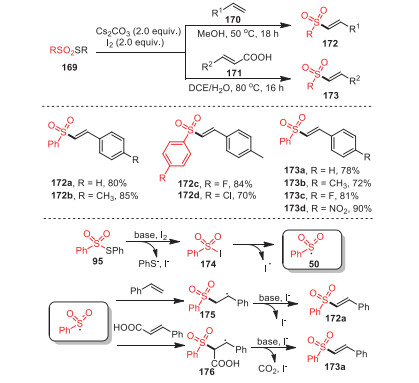
A simple method to construct various vinyl sulfones 172 by sulfonylation of thiosulfonates 169 with styrenes 170 or cinnamic acid derivatives 171 was discovered by Jang's group in 2018 (Scheme 25) [110]. Notably, this sulfonylation appeared to be more functional for electron-deficient thiosulfonates and electron-rich styrene derivatives. In addition, under standard conditions, electron-deficient cinnamic acid derivatives could react with phenyl thiosulfonates smoothly, providing corresponding products in moderate to excellent yields. The strategy was characterized by mild conditions, environmental friendliness, and excellent functional group tolerance.

|
Download:
|
| Scheme 25. The synthesis of vinyl sulfones mediated by iodine. | |
{kind=link}
Some control experiments indicated that the above process may involve a radical pathway, and a rational mechanism was outlined as Scheme 25. First, sulfonyl iodide 174, derived from the reaction of thiosulfonate with I2, underwent homolytic cleavage to form sulfonyl radical 50, which was then added to either styrene or cinnamic acid. Finally, the radical intermediates 175 or 176 performed base-assisted elimination of HI or HI and CO2, resulting in vinyl sulfone 172a or 173a, respectively.
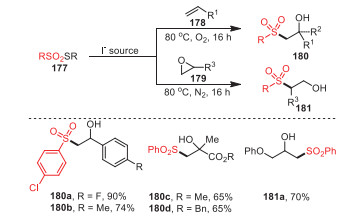
In the same year, the same group also studied the reactivity of thiosulfonates 177, which could selectively generate β-hydroxylsulfones with moderate to excellent yields under the condition of organic base KI and an aqueous solvent (Scheme 26) [111]. Obviously, this reaction could occur smoothly between either electron-withdrawing or electron-donating thiosulfonates and substituted styrenes, and the desired products were obtained successfully. In addition, electron-deficient alkenes, including methyl methacrylate and benzyl methacrylate, were obtained, with 65% yields of 180c and 180d, respectively.

|
Download:
|
| Scheme 26. The synthesis of diverse β-hydroxysulfones under metal-free conditions. | |
{kind=link}
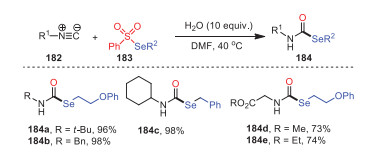
In 2019, Ji, Wang and coworkers developed an effective method for the construction of secondary selenocarbamates 184 from isocyanides 182, selenosulfonates 183, and water by a radical-chain reaction under metal-free conditions (Scheme 27) [112]. This approach was characterized by simple operation, mild conditions, no need for additional catalysts, and good functional group tolerance. Obviously, either alkyl- or aryl-substituted isocyanides reacted smoothly under standard conditions and yielded corresponding products with considerable yields. This showed that the universality of the current process applied to both chain and ring compounds. Surprisingly, the primary isocyanides 184d and 184e target products were also smoothly obtained, with yields of 73% and 74%, respectively.

|
Download:
|
| Scheme 27. The synthesis of selenocarbamates under metal-free conditions. | |
{kind=link}
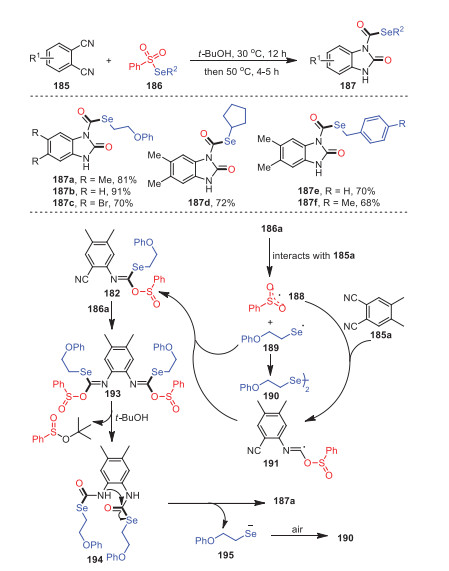
Later, a method of constructing 2-benzimidazolone derivatives 187 using the metal-free reaction of ortho-diisocyanoarenes 185 and selenosulfonates 186 was first developed by the same group (Scheme 28) [113]. The reaction was easy to handle and proceeded smoothly with moderate to excellent yields and tolerance of functional groups. Methyl- and bromide-substituted isocyanides all reacted successfully under standard conditions to obtain the target products with 81% and 70% yields, respectively. Furthermore, not only aryl-substituted selenosulfonates were suitable for this process, but cycloalkane-substituted selenosulfonates also had good tolerance under the same conditions. Based on the results of relevant control experiments and previous literature reports, a possible mechanism involving a radical process was described, as shown in Scheme 28. Herein, the formation of radical intermediate 188 was due to the intermolecular interaction between 185a and 186a.

|
Download:
|
| Scheme 28. The synthesis of N-(carboselenoate) benzimidazolones under metal-free conditions. | |
{kind=link}
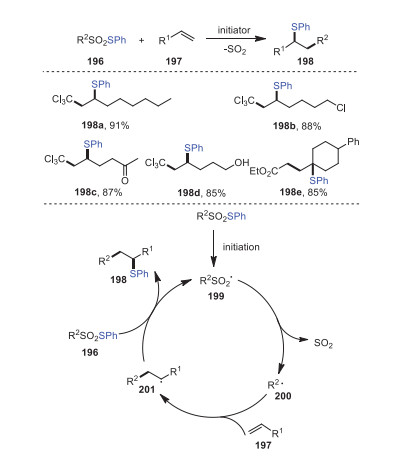
In 2020, Renaud's group introduced an efficient method for the thioalkylation of olefins by radical desulfitation sulfur-group transfer based on the use of readily available thiosulfonates 196 as starting materials (Scheme 29) [114]. Using Cl3CSO2SPh as a starting material and diauryl peroxide (DLP) as an initiator, the desulfitative thioalkylation of 1-octene 197a was attempted, and a 91% yield of corresponding product 198a was successfully obtained. A considerable number of functional groups such as chlorines, ketones, and even alcohols could be well tolerated by this process. Mechanistically, a sulfonyl radical 199 was generated as a result of DLP, light, or V-40 initiation processes, which underwent a rapid desulfitation process resulting in an electrophilic radical 200 being added to the olefin 197. The radical intermediate 201 then reacted with thiosulfinate to form a thioalkylated alkene.

|
Download:
|
| Scheme 29. Desulfitative thioalkylation of alkenes under metal-free conditions. | |
{kind=link}
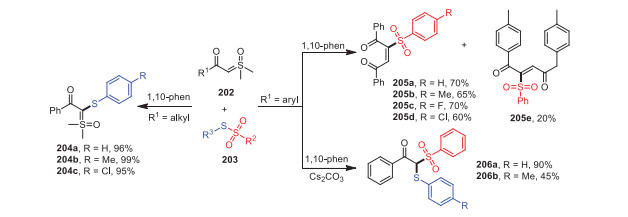
In 2020, Wang and coworkers established a radical cascade reaction between sulfoxonium ylides 202 and thiosulfonates 203 in the absence of transition metals and smoothly selectively prepared 1, 4-diketone compounds 204, arylthiosulfoxide-ylides 205, and β-keto thiosulfones 206 (Scheme 30) [115]. Notably, the desired products could be obtained successfully in moderate to excellent yields when using S-phenyl arylsulfonates possessing electron-withdrawing or electron-donating groups as substrates. However, when 2-(dimethyl(oxo)-λ6-sulfanylidene)-1-(p-tolyl)ethan-1-one 202e was used as a starting material, the yield decreased to 20%. Mechanistic studies confirmed that this transformation was a free-radical reaction process, and 1, 10-phenanthroline could stabilize free radicals in this route.

|
Download:
|
| Scheme 30. The preparation of 1, 4-diketone compounds, arylthiosulfoxide-ylides, and β-keto thiosulfones under metal-free conditions. | |
{kind=link}
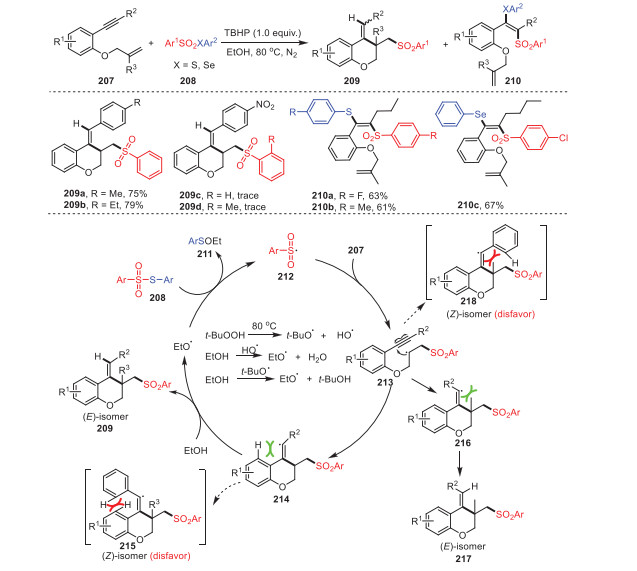
A method to successfully construct (E)−3-((arylsulfonyl)-methyl)-4-substituted benzylidenechromene derivatives 209 through radical cyclization of oxygen-containing 1, 7-enynes 207 and thiosulfonates 208 with the assistance of TBHP was proposed by Wang, Rong and coworkers. (Scheme 31) [116]. Remarkably, different alkyl groups such as methyl as well as ethyl on the aromatic ring at the terminal alkyne (R2) could participate under standard conditions smoothly, obtaining corresponding products 209a and 209b with moderate yields. However, 1, 7-enynes bearing a stronger electron-withdrawing group (NO2) were not suitable for this reaction, indicating that the substituent properties of 1, 7-enynes had a great impact on the reaction. The gram-scale reaction of this procedure was also verified, and the yield was good. A feasible mechanism was reported, as shown in Scheme 31, based on the results of control experiments.

|
Download:
|
| Scheme 31. The construction of (E)-3-((arylsulfonyl)-methyl)-4-substituted benzylidenechromene derivatives under metal-free conditions. | |
{kind=link}
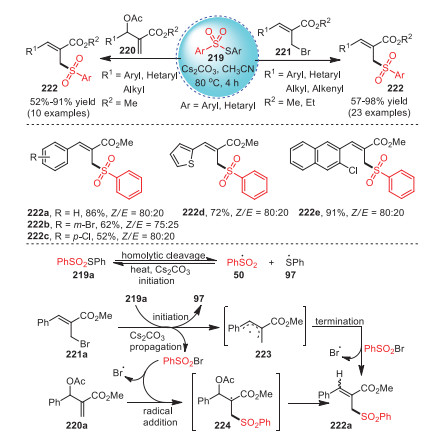
In 2021, Reddy's group proposed a method for the synthesis of allyl sulfones 222 with high yields and stereoselectivity by radical sulfonation reaction of Morita–Bayis–Hillman adducts and thiosulfonates 219 promoted by Cs2CO3 (Scheme 32) [117]. Surprisingly, aryl- and heteraryl-derived substrates could also tolerate this method, and the target products were successfully obtained with moderate to excellent yields. In addition, these allyl sulfones, especially allyl (hetero)aryl sulfones, had anticancer and abnormal cell-proliferation activities.

|
Download:
|
| Scheme 32. The synthesis of allyl sulfones mediated by Cs2CO3. | |
{kind=link}
Mechanistically, at the very start, in the presence of Cs2CO3, PhSSO2Ph underwent homolytic cleavage resulting in the production of sulfonyl radical 50 and thiyl radical 97. Afterwards, the propagation of 221a explained the formation of allyl radical 223 and termination product sulfonyl bromide (PhSO2Br). In the end, the desired allyl sulfone 222a was obtained via the sulfonylation of product 223 and PhSO2Br. Similarly, the radical addition of substrate 220a with sulfonyl radical generated radical 224, which underwent elimination to access the desired allyl sulfone 222a.
4. Conclusions and outlookIn this review, the recent progress in the synthesis and applications of thiosulfonates and selenosulfonates as radical reagents is reviewed, and the reaction scopes, limitations, and some mechanisms are also discussed. Depending on the reaction system, metal catalysis, visible-light catalysis, synergistic catalysis, and other types including azo compounds, heat, organic molecules, or peroxide-induced radical reactions were classified. After the above discussion, it can be found that thiosulfonates and selenosulfonates can utilize these methods to provide a stable and powerful platform for the efficient construction of various selenium- and sulfur-containing heterocyclic compounds and difunctional products. The successful examples described in this review convincingly demonstrate the high potential of thiosulfonates and selenosulfonates for drug discovery and applications. Among these synthesis methods, photochemistry and synergistic metal/photoredox catalysis techniques provide a more environmentally friendly and sustainable alternative for the applications of thiosulfonates and selenosulfonates and illustrate the development prospects of chalcogenative sulfones as free radicals.
Although significant progress has been made in this field over the last few years, several issues and challenges remain in fully developing the potential applications of thiosulfonates and selenosulfonates, which should be further exploited and improved. First, more catalytic means should be explored in the applications of thiosulfonates and selenosulfonates as free-radical reagents, especially electrochemical synthesis methods, since electrochemical strategies have become increasingly powerful tools for the synthesis of organic compounds in recent years. Second, migration cyclization strategies involving thiosulfonates and selenosulfonates as radical reagents should be established to construct more complex polycyclic compounds, since polycyclic frameworks play an important role in medicinal chemistry and organic materials. In addition, the harmful consequences of metal catalysts and peroxides used in the reaction process cannot be ignored. Hopefully this paper will encourage more researchers to contribute to this emerging field by developing more environmentally friendly catalytic systems.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21801007), Qingchuang Technology Support Program of University in Shandong Province (No. 2021KJ066) and Hunan Engineering Laboratory for analyse and Drugs Development of Ethnomedicine in Wunlin Mountains (No. Hgxy2103).
| [1] |
T. Kondo, T. Mitsudo, Chem. Rev. 100 (2000) 3205-3220. DOI:10.1021/cr9902749 |
| [2] |
M. Mellah, A. Voituriez, E. Schulz, Chem. Rev. 107 (2007) 5133-5209. DOI:10.1021/cr068440h |
| [3] |
J.E. Taylor, S.D. Bull, J.M.J. Williams, Chem. Soc. Rev. 41 (2012) 2109-2121. DOI:10.1039/c2cs15288f |
| [4] |
B. Mandal, B. Basu, RSC Adv. 4 (2014) 13854-13881. DOI:10.1039/c3ra45997g |
| [5] |
E.A. Ilardi, E. Vitaku, J.T. Njardarson, J. Med. Chem. 57 (2014) 2832-2842. DOI:10.1021/jm401375q |
| [6] |
C. Ni, M. Hu, J. Hu, Chem. Rev. 115 (2015) 765-825. DOI:10.1021/cr5002386 |
| [7] |
D. Wang, P. Cao, B. Wang, et al., Org. Lett. 17 (2015) 2420-2423. DOI:10.1021/acs.orglett.5b00934 |
| [8] |
X.X. Shao, C.F. Xu, L. Lu, Q.L. Shen, Acc. Chem. Res. 48 (2015) 1227-1236. DOI:10.1021/acs.accounts.5b00047 |
| [9] |
M.H. Feng, B.Q. Tang, S.H. Liang, X.F. Jiang, Curr. Top. Med. Chem. 16 (2016) 1200-1216. DOI:10.2174/1568026615666150915111741 |
| [10] |
X. Xiao, M. Feng, X. Jiang, Angew. Chem. Int. Ed. 55 (2016) 14121-14125. DOI:10.1002/anie.201608011 |
| [11] |
Y. Li, M. Wang, X. Jiang, ACS Catal. 7 (2017) 7587-7592. DOI:10.1021/acscatal.7b02735 |
| [12] |
C. Fortugno, G. Varchi, A. Guerrini, et al., Biomed. Anal. 95 (2014) 151-157. DOI:10.1016/j.jpba.2014.03.002 |
| [13] |
G. Mugesh, W.W. du Mont, H. Sies, Chem. Rev. 101 (2001) 2125-2180. DOI:10.1021/cr000426w |
| [14] |
C.W. Nogueira, G. Zeni, J.B.T. Rocha, Chem. Rev. 104 (2004) 6255-6286. DOI:10.1021/cr0406559 |
| [15] |
J.J. Ai, J. Li, S.J. Ji, S.Y. Wang, Chin. Chem. Lett. 32 (2021) 721-724. DOI:10.1016/j.cclet.2020.07.007 |
| [16] |
X.Z. Li, P. Liu, J. He, et al., Green Synth. Catal. 2 (2021) 381-384. DOI:10.1016/j.gresc.2021.08.006 |
| [17] |
O. Foss, J. Am. Chem. Soc. 69 (1947) 2236-2237. |
| [18] |
B.M. Trost, Chem. Rev. 78 (1978) 363-382. DOI:10.1021/cr60314a002 |
| [19] |
D.H.R. Barton, B. Lacher, B. Misterkiewics, S.Z. Zard, Tetrahedron 44 (1988) 1153-1158. DOI:10.1016/S0040-4020(01)85895-6 |
| [20] |
K. Fujiki, E. Yoshida, Synth. Commun. 29 (1999) 3289-3294. DOI:10.1080/00397919908085956 |
| [21] |
S. Kim, S. Kim, N. Otsuka, I. Ryu, Angew. Chem. Int. Ed. 44 (2005) 6183-6186. DOI:10.1002/anie.200501606 |
| [22] |
F. Kopp, P. Knochel, Org. Lett. 9 (2007) 1639-1641. DOI:10.1021/ol063136w |
| [23] |
S.H. Wunderlich, P. Knochel, Angew. Chem. Int. Ed. 46 (2007) 7685-7688. DOI:10.1002/anie.200701984 |
| [24] |
V. Girijavallabhan, C. Alvarez, F.G. Njoroge, J. Org. Chem. 76 (2011) 6442-6446. DOI:10.1021/jo201016z |
| [25] |
P. Saravanan, P. Anbarasan, Org. Lett. 16 (2014) 848-851. DOI:10.1021/ol4036209 |
| [26] |
S. Yoshida, Y. Sugimura, Y. Hazamam, et al., Chem. Commun. 51 (2015) 16613-16616. DOI:10.1039/C5CC07463K |
| [27] |
P.K. Shyam, H.Y. Jang, J. Org. Chem. 82 (2017) 1761-1767. DOI:10.1021/acs.joc.6b03016 |
| [28] |
N. Wang, P. Saidhareddy, X. Jiang, Nat. Prod. Rep. 37 (2020) 246-275. DOI:10.1039/C8NP00093J |
| [29] |
H. Haruki, M.G. Pedersen, K.I. Gorska, F. Pojer, K. Johnsson, Science 340 (2013) 987-991. DOI:10.1126/science.1232972 |
| [30] |
T.J. Deming, Bioconjugate Chem. 28 (2017) 691-700. DOI:10.1021/acs.bioconjchem.6b00696 |
| [31] |
M. Yan, J.C. Lo, J.T. Edwards, P.S. Baran, J. Am. Chem. Soc. 138 (2016) 12692-12714. DOI:10.1021/jacs.6b08856 |
| [32] |
S. Crespi, M. Chem. Rev. 120 (2020) 9790-9833. DOI:10.1021/acs.chemrev.0c00278 |
| [33] |
A. Studer, D.P. Curran, Angew. Chem. Int. Ed. 55 (2016) 58-102. DOI:10.1002/anie.201505090 |
| [34] |
K. Sun, Z.D. Shi, Z.H. Liu, et al., Org. Lett. 20 (2018) 6687-6690. DOI:10.1021/acs.orglett.8b02733 |
| [35] |
K. Sun, S.N. Wang, R.R. Feng, et al., Org. Lett. 21 (2019) 2052-2055. DOI:10.1021/acs.orglett.9b00240 |
| [36] |
K. Sun, G.F. Li, Y.Y. Li, et al., Adv. Synth. Catal. 362 (2020) 1947-1954. DOI:10.1002/adsc.202000040 |
| [37] |
X. Wang, Q.L. Wang, Y.R. Xue, et al., Chem. Commun. 56 (2020) 4436-4439. DOI:10.1039/D0CC01079K |
| [38] |
K. Sun, X. Wang, C. Li, H. Wang, L. Li, Org. Chem. Front. 7 (2020) 3100-3119. DOI:10.1039/D0QO00849D |
| [39] |
X. Wang, S. Guo, Y. Zhang, et al., Adv. Synth. Catal. 363 (2021) 3290-3296. DOI:10.1002/adsc.202100208 |
| [40] |
Y. Xing, C. Li, J.P. Meng, et al., Adv. Synth. Catal. 363 (2021) 3913-3936. DOI:10.1002/adsc.202100446 |
| [41] |
X. Wang, Y. Zhang, K. Sun, J.P. Meng, B. Zhang, Chin. J. Org. Chem. 41 (2021) 4588-4609. DOI:10.6023/cjoc202109046 |
| [42] |
S. Guo, X. Wang, D.Y. Zhao, et al., Asian J. Org. Chem. 11 (2022). |
| [43] |
X. Wang, J. Lei, S. Guo, et al., Chem. Commun. 58 (2022) 1526-1529. DOI:10.1039/D1CC06323E |
| [44] |
C. Ghiazza, T. Billard, Eur. J. Org. Chem. 2021 (2021) 5571-5584. DOI:10.1002/ejoc.202100944 |
| [45] |
S. Huang, Z.H. Xia, K. Lu, et al., Chin. J. Chem. 38 (2020) 1625-1628. DOI:10.1002/cjoc.202000279 |
| [46] |
P. Mampuys, C.R. McElroy, J.H. Clark, R.V.A. Orru, B.U.W. Maes, Adv. Synth. Catal. 362 (2020) 3-64. DOI:10.1002/adsc.201900864 |
| [47] |
M.D. Bentley, I.B. Douglass, J.A. Lacadie, J. Org. Chem. 37 (1972) 333-334. DOI:10.1021/jo00967a040 |
| [48] |
G.G. Liang, J. Chen, J.L. Chen, et al., Tetrahedron Lett. 53 (2012) 6768-6770. DOI:10.1016/j.tetlet.2012.09.132 |
| [49] |
N. Taniguchi, Eur. J. Org. Chem. 2014 (2014) 5691-5694. DOI:10.1002/ejoc.201402847 |
| [50] |
P. Natarajan, Tetrahedron Lett. 56 (2015) 4131-4134. DOI:10.1016/j.tetlet.2015.05.050 |
| [51] |
Y. Zheng, F.L. Qing, Y. Huang, X.H. Xu, Adv. Synth. Catal. 358 (2016) 3477-3481. DOI:10.1002/adsc.201600633 |
| [52] |
X.J. Li, C. Zhou, P.H. Diao, Y.Q. Ge, C. Guo, Tetrahedron Lett. 58 (2017) 1296-1300. DOI:10.1016/j.tetlet.2017.02.042 |
| [53] |
G.Y. Zhang, S.S. Lv, A. Shoberu, J.P. Zou, J. Org. Chem. 82 (2017) 9801-9807. DOI:10.1021/acs.joc.7b01121 |
| [54] |
Z.Z. Yang, Y.S. Shi, Z. Zhan, et al., ChemElectroChem 5 (2018) 3619-3623. DOI:10.1002/celc.201801058 |
| [55] |
X. Zhang, T. Cui, Y. Zhang, et al., Adv. Synth. Catal. 361 (2019) 2014-2019. DOI:10.1002/adsc.201900047 |
| [56] |
A.K. Pandey, A. Kumar, N. Verma, S.K. Srivastava, Beilstein Arch (2021) 202115. |
| [57] |
X.C. Wang, C.M. Zhang, Y.D. Zhang, Z.H. Ren, Z.H. Guan, Chin. J. Org. Chem. 40 (2020) 1618-1624. DOI:10.6023/cjoc202002009 |
| [58] |
H. Li, Y.X. Han, Z. Yang, et al., Chin. Chem. Lett. 32 (2021) 1709-1712. DOI:10.1016/j.cclet.2020.12.027 |
| [59] |
P. Zhang, W.J. Chang, H.Y. Jiao, et al., Chin. Chem. Lett. 32 (2021) 1717-1720. DOI:10.1016/j.cclet.2021.01.024 |
| [60] |
Z. Zhang, W.X. Chang, Chin. J. Org. Chem. 41 (2021) 1835-1850. |
| [61] |
S.P. Wu, D.K. Wang, Q.Q. Kang, et al., Chem. Commun. 57 (2021) 8288-8291. DOI:10.1039/D1CC03252F |
| [62] |
Y. Liu, S.Y. Xing, J. Zhang, et al., Org. Chem. Front. 9 (2022) 1375-1382. DOI:10.1039/D1QO01873F |
| [63] |
Y.H. Lv, J.P. Meng, C. Li, et al., Adv. Synth. Catal. 363 (2021) 5235-5265. DOI:10.1002/adsc.202101184 |
| [64] |
K. Sun, Y. Li, Q. Zhang, Sci. China Chem. 58 (2015) 1354-1358. DOI:10.1007/s11426-015-5385-y |
| [65] |
K. Sun, X. Wang, L.L. Liu, et al., ACS Catal. 5 (2015) 7194-7198. DOI:10.1021/acscatal.5b02411 |
| [66] |
D.H. Zhu, X.X. Shao, X. Hong, L. Lu, Q.L. Shen, Angew. Chem. 128 (2016) 16039-16043. DOI:10.1002/ange.201609468 |
| [67] |
P.K. Shyam, S. Son, H.Y. Jang, Eur. J. Org. Chem. 2017 (2017) 5025-5031. DOI:10.1002/ejoc.201700971 |
| [68] |
S. Huang, N. Thirupathi, C.H. Tung, Z.H. Xu, J. Org. Chem. 83 (2018) 9449-9455. DOI:10.1021/acs.joc.8b01161 |
| [69] |
Q.J. Liang, P.J. Walsh, T.Z. Jia, ACS Catal. 10 (2020) 2633-2639. DOI:10.1021/acscatal.9b04887 |
| [70] |
C.K. Prier, D.A. Rankic, D.W. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [71] |
M.N. Hopkinson, A. Tlahuext-Aca, F. Glorius, Acc. Chem. Res. 49 (2016) 2261-2272. DOI:10.1021/acs.accounts.6b00351 |
| [72] |
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075-10166. DOI:10.1021/acs.chemrev.6b00057 |
| [73] |
X.Y. Yu, J.R. Chen, W.J. Xiao, Chem. Rev. 121 (2020) 506-561. |
| [74] |
T. Rawner, E. Lutsker, C.A. Kaiser, O. Reiser, ACS Catal. 8 (2018) 3950-3956. DOI:10.1021/acscatal.8b00847 |
| [75] |
B. Lipp, L.M. Kammer, M. Kücükdisli, et al., Chem. Eur. J. 25 (2019) 8965-8969. DOI:10.1002/chem.201901175 |
| [76] |
L.M. Kammer, M. Krumb, B. Spitzbarth, et al., Org. Lett. 22 (2020) 3318-3322. DOI:10.1021/acs.orglett.0c00614 |
| [77] |
J.K. Liu, H. Yao, X.N. Li, et al., Org. Chem. Front. 7 (2020) 1314-1320. DOI:10.1039/D0QO00343C |
| [78] |
J. Li, X.E. Yang, S.L. Wang, et al., Org. Lett. 22 (2020) 4908-4913. DOI:10.1021/acs.orglett.0c01776 |
| [79] |
K. Gadde, P. Mampuys, A. Guidetti, et al., ACS Catal. 10 (2020) 8765-8779. DOI:10.1021/acscatal.0c02159 |
| [80] |
C.M. Huang, J. Li, J.J. Ai, et al., Org. Lett. 22 (2020) 9128-9132. DOI:10.1021/acs.orglett.0c03562 |
| [81] |
Y. Dong, P. Ji, Y.T. Zhang, et al., Org. Lett. 22 (2020) 9562-9567. DOI:10.1021/acs.orglett.0c03624 |
| [82] |
H. Chen, Y.Y. Yan, N.N. Zhang, et al., Org. Lett. 23 (2021) 376-381. DOI:10.1021/acs.orglett.0c03876 |
| [83] |
F. Wang, S.Y. Wang, Org. Chem. Front. 8 (2021) 1976-1982. DOI:10.1039/D1QO00085C |
| [84] |
W.Y. Li, L. Zhou, Green Chem. 23 (2021) 6652-6658. DOI:10.1039/D1GC02036F |
| [85] |
W.Z. Bi, W.J. Zhang, Z.J. Li, et al., Org. Biomol. Chem. 19 (2021) 8701-8705. DOI:10.1039/D1OB01592C |
| [86] |
X.Y. Liu, S.Y. Tian, Y.F. Jiang, W.D. Rao, S.Y. Wang, Org. Lett. 23 (2021) 8246-8251. DOI:10.1021/acs.orglett.1c02981 |
| [87] |
Y. Liu, N.N. Zhang, Y.L. Xu, Y.Y. Chen, J. Org. Chem. 86 (2021) 16882-16891. DOI:10.1021/acs.joc.1c02082 |
| [88] |
J. Xuan, Z. Zhang, W. Xiao, Angew. Chem. Int. Ed. 54 (2015) 15632-15641. DOI:10.1002/anie.201505731 |
| [89] |
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [90] |
J. Xuan, W. Xiao, Angew. Chem. Int. Ed. 51 (2012) 6828-6838. DOI:10.1002/anie.201200223 |
| [91] |
J.M.R. Narayanam, C.R.J. Stephenson, Chem. Soc. Rev. 40 (2011) 102-113. DOI:10.1039/B913880N |
| [92] |
T.P. Yoon, M.A. Ischay, J. Du, Nat. Chem. 2 (2010) 527-532. DOI:10.1038/nchem.687 |
| [93] |
T. Chatterjee, N. Iqbal, Y. You, E.J. Cho, Acc. Chem. Res. 49 (2016) 2284-2294. DOI:10.1021/acs.accounts.6b00248 |
| [94] |
K.L. Skubi, T.R. Blum, T.P. Yoon, Chem. Rev. 116 (2016) 10035-10074. DOI:10.1021/acs.chemrev.6b00018 |
| [95] |
D. Kalyani, K.B. McMurtrey, S.R. Neufeldt, M.S. Sanford, J. Am. Chem. Soc. 133 (2011) 18566-18569. DOI:10.1021/ja208068w |
| [96] |
S.R. Neufeldt, M.S. Sanford, Adv. Synth. Catal. 354 (2012) 3517-3522. DOI:10.1002/adsc.201200738 |
| [97] |
J. Zoller, D.C. Fabry, M.A. Ronge, M. Rueping, Angew. Chem. Int. Ed. 53 (2014) 13264-13268. DOI:10.1002/anie.201405478 |
| [98] |
J. Xuan, T. Zeng, Z. Feng, et al., Angew. Chem. Int. Ed. 54 (2015) 1625-1628. DOI:10.1002/anie.201409999 |
| [99] |
J.A. Terrett, J.D. Cuthbertson, V.W. Shurtleff, D.W.C. MacMillan, Nature 524 (2015) 330-334. DOI:10.1038/nature14875 |
| [100] |
Z. Zuo, H. Cong, W. Li, et al., J. Am. Chem. Soc. 138 (2016) 1832-1835. DOI:10.1021/jacs.5b13211 |
| [101] |
Y. Ye, M.S. Sanford, J. Am. Chem. Soc. 134 (2012) 9034-9037. DOI:10.1021/ja301553c |
| [102] |
A.Y. Chan, I.B. Perry, N.B. Bissonnette, et al., Chem. Rev. 122 (2022) 1485-1542. DOI:10.1021/acs.chemrev.1c00383 |
| [103] |
H.Y. Li, C.C. Shan, C.H. Tung, Z.H. Xu, Chem. Sci. 8 (2017) 2610-2615. DOI:10.1039/C6SC05093J |
| [104] |
T.T. Song, H.Y. Li, F. Wei, C.H. Tung, Z.H. Xu, Tetrahedron Lett. 60 (2019) 916-919. DOI:10.1016/j.tetlet.2019.02.039 |
| [105] |
R. Zhang, P. Xu, S.Y. Wang, S.J. Ji, J. Org. Chem. 84 (2019) 12324-12333. DOI:10.1021/acs.joc.9b01626 |
| [106] |
X. Zhou, Z.Y. Peng, P.G. Wang, Q.C. Liu, T.Z. Jia, Org. Lett. 23 (2021) 1054-1059. DOI:10.1021/acs.orglett.0c04254 |
| [107] |
T.G. Back, S. Collins, R.G. Kerr, J. Org. Chem. 48 (1983) 3077-3084. DOI:10.1021/jo00166a030 |
| [108] |
P. Mampuys, Y.P. Zhu, S. Sergeyev, et al., Org. Lett. 18 (2016) 2808-2811. DOI:10.1021/acs.orglett.6b01023 |
| [109] |
N. Taniguchi, Tetrahedron 73 (2017) 2030-2035. DOI:10.1016/j.tet.2017.02.047 |
| [110] |
S.J. Hwang, P.K. Shyam, H.Y. Jang, Bull. Korean Chem. Soc. 39 (2018) 535-539. DOI:10.1002/bkcs.11426 |
| [111] |
S. Son, P.K. Shyam, H. Park, I. Jeong, H.Y. Jang, Eur. J. Org. Chem. 2018 (2018) 3365-3371. DOI:10.1002/ejoc.201800778 |
| [112] |
Y. Fang, C. Liu, F. Wang, et al., Org. Chem. Front. 6 (2019) 660-663. |
| [113] |
Y. Fang, C. Liu, W.D. Rao, S.Y. Wang, S.J. Ji, Org. Lett. 21 (2019) 7687-7691. DOI:10.1021/acs.orglett.9b01886 |
| [114] |
L. Cao, C. Jimeno, P. Renaud, Adv. Synth. Catal. 362 (2020) 3644-3648. DOI:10.1002/adsc.202000657 |
| [115] |
F. Wang, B.X. Liu, W.D. Rao, S.Y. Wang, Org. Lett. 22 (2020) 6600-6604. DOI:10.1021/acs.orglett.0c02370 |
| [116] |
K.M. Mao, M.W. Bian, L. Dai, et al., Org. Lett. 23 (2021) 218-224. DOI:10.1021/acs.orglett.0c03946 |
| [117] |
A. Shankar, M. Waheed, R.J. Reddy, SynOpen 5 (2021) 91-99. DOI:10.1055/a-1422-9411 |