 2023, Vol. 34
2023, Vol. 34 


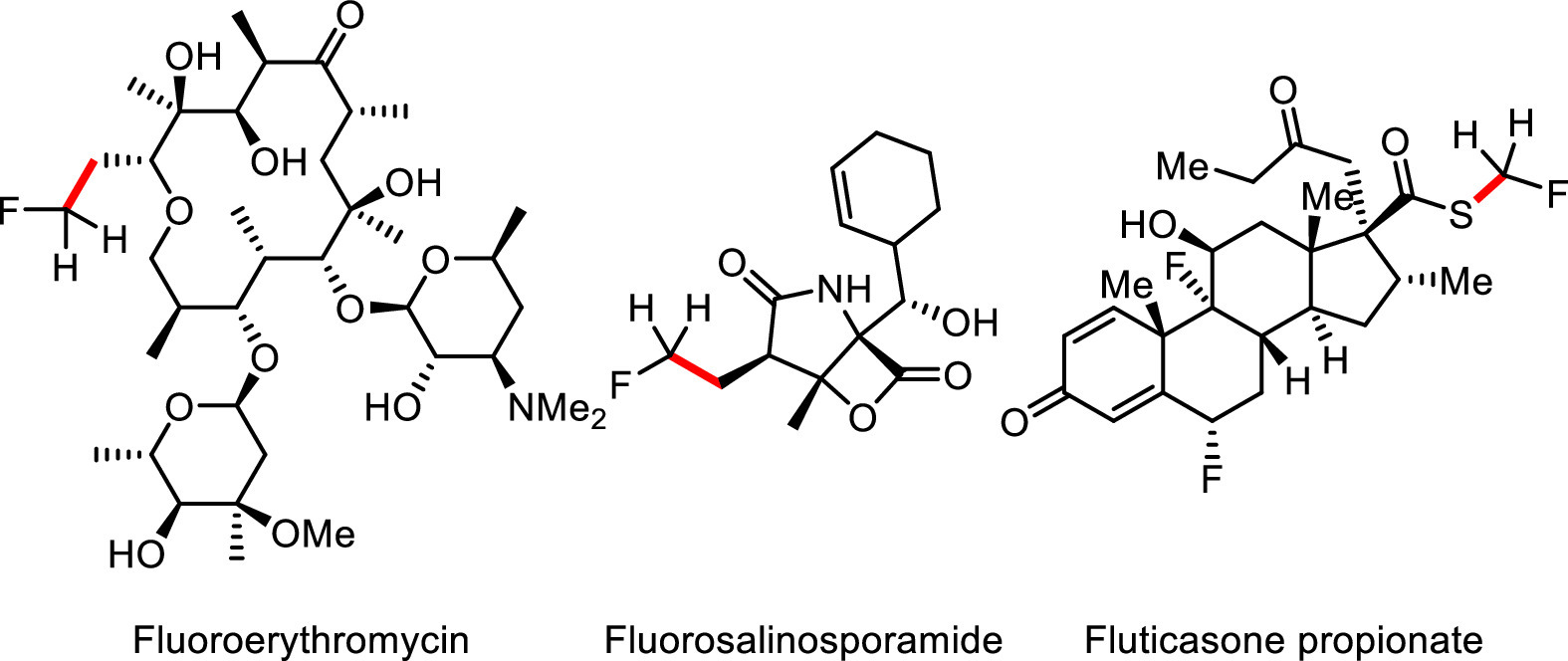
Monofluorinated compounds serve as the most important kind of fluorine-containing molecules in pharmaceuticals and agrochemicals, due to their significantly increased lipid solubility, permeability, metabolic stability and bioactivity by comparison of the parent molecules (Fig. 1) [1-5]. Accordingly, the selective incorporation of fluorine atom into organic molecules has sustained significant interest in recent years [6-11], and persistent efforts have been devoted to establishing efficient methods by direct fluorination or indirect monofluoroalkylation to make monofluorinated alkanes. Indeed, the direct C-F bond construction from alkyl alcohols was first reported in the 1970s and was later popularized and expanded to other alkyl-X (X = halide, COOH, etc.) through decades of hard work from lots of groups [12-15]. Even though a number of fluorinating agents have been developed for direct fluorination in the past decades [16-28], the known methods for synthesis of alkyl fluorides, especially terminal aliphatic fluorides, were still suffered from limited substrate compatibility, low catalytic reactivity and poor site selectivity. Considering the C-F bond forming reductive elimination from the metal fluorides was quite difficult, an alternative C—C cross-coupling strategy offered a solution for synthesis of primary alkyl fluorides via a transition-metal catalyzed monofluoromethylation of aryl/alkyl halides or boronic acids [29-35]. Recently, our group reported a nickel-catalyzed reductive cross-coupling [32-34] of alkyl (pesudo)halides and fluoromethyl bromide to access various aliphatic fluorides, in which the reaction was initiated by oxidation addition of CH2FBr to Ni0 and followed by in situ reduction of NiII to NiI species for further activation of alkyl halides to furnish the final primary fluorides (Scheme 1a).

|
Download:
|
| Fig. 1. Bioactive molecules containing terminal fluorine atom. | |

|
Download:
|
| Scheme 1. Synthesis of primary alkyl fluorides via nickel-catalyzed C—C cross-coupling. | |
As the most abundant, low-cost and versatile feedstock on large scale readily available from petrochemical industry, simple unactivated alkenes have been widely utilied to a great variety of organic transformations to produce various fine chemicals or useful molecules for industrial production and academic research [36-38]. In view of the normally used electrophiles and nucleophiles were typically generated from alkenes, no doubtedly, the direct use of alkenes as the coupling partner for nickel-catalyzed monofluoroalkylation represents the most step-economic and efficient way to make primary aliphatic fluorides [39, 40]. As a powerful synthetic strategy developed rapidly in the past decade, the use of simple unactivated alkenes as alkyl organometallic equivalents in transition-metal catalzyed cross-coupling reactions, in which silanes were normally used as hydride sources to generate alkyl organic metal species by insertion of C═C double bond to M-H species generated in situ [41-44]. As one part of our continuous interests on monofluoroalkylation, we speculated that such alkylated NiI species could activate monofluoromethyl halide via two SET steps, affording the terminal alkyl fluorides after the following reductive elimination.
Herein, we report the first example of nickel-catalyzed three-component hydrofluoromethylation of unactivated alkenes with industrial raw fluoroiodomethane as the fluoromethylating source, which furnished diverse terminal aliphatic fluorides in a convenient and efficient manner (Scheme 1b). This transformation tolerates diverse collection of terminal alkenes and demonstrates mild conditions, high catalytic reactivity and excellent functional group compatibility. Mechanistic studies support a Ni-H initiated hydronickelation of alkenes followed by monofluoromethyl radical-involved SET oxidative addition.
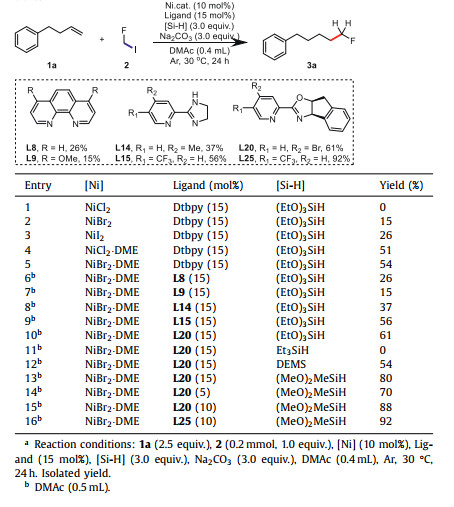
Our study commenced with but-3-en-1-ylbenzene (1a) as the pilot substrate, (EtO)3SiH as the hydrogen source, fluoroiodomethane (2) as the coupling partner, Na2CO3 (3.0 equiv.) as the base in DMAc. By using dtbpy (15 mol%) as the ligand, a careful investigation of nickel sources were firstly performed (Table 1, entries 1–5; for details, see Supporting information). While NiCl2 afforded none of the desired aliphatic fluoride 3a (Table 1, entry 1), to our interests, NiBr2 and NiI2 furnished 3a successfully under the same conditions, albeit with low yields at 15% or 26% respectively (Table 1, entries 2 and 3). In view of the solubility of different nickel sources may play a key role for the catalytic efficience, easily soluble nickels like NiCl2·DME and NiBr2·DME were next explored in this catalytic system to furnish the desired 3a in relatively higher yield in 51% and 54% yields respectively. To further improve the yield of this transformation, diverse nitrogen ligands have been carefully investigated. While phenanthroline and its derivatives (L8 and L9) afforded 3a with only lower yields, pyridine-imidazoline (L15) gave a slightly higer yield at 56%. As sterically hindered ligands could facilitate the reductive elimination of this nickel catalytic cycle, pyridine-oxazoline ligands were next examined and L20 could further prove the yield to 61% (Table 1, entry 10). Considering the important role of external hydrogen source played in forming the Ni-H species, several hydrogen sources were then examined (Table 1, entries 11–13), among which (MeO)2MeSiH increased productivity to 80%. Meanwhile, screening of the ratio of nickel salt to ligand revealed that 1:1 works best (Table 1, entries 14 and 15). To our satisfactory, a detailed examination of pyridine-oxazoline ligand analogs (for details see Supporting information) indicated that L25 with a CF3 group installed on the pyridine ring proved to be the best choice (92% yield, Table 1, entry 16).
|
|
Table 1 Nickel-catalyzed hydromonofluoromethylation of alkenes: optimization of conditions.a |
{kind=link}
{kind=link}
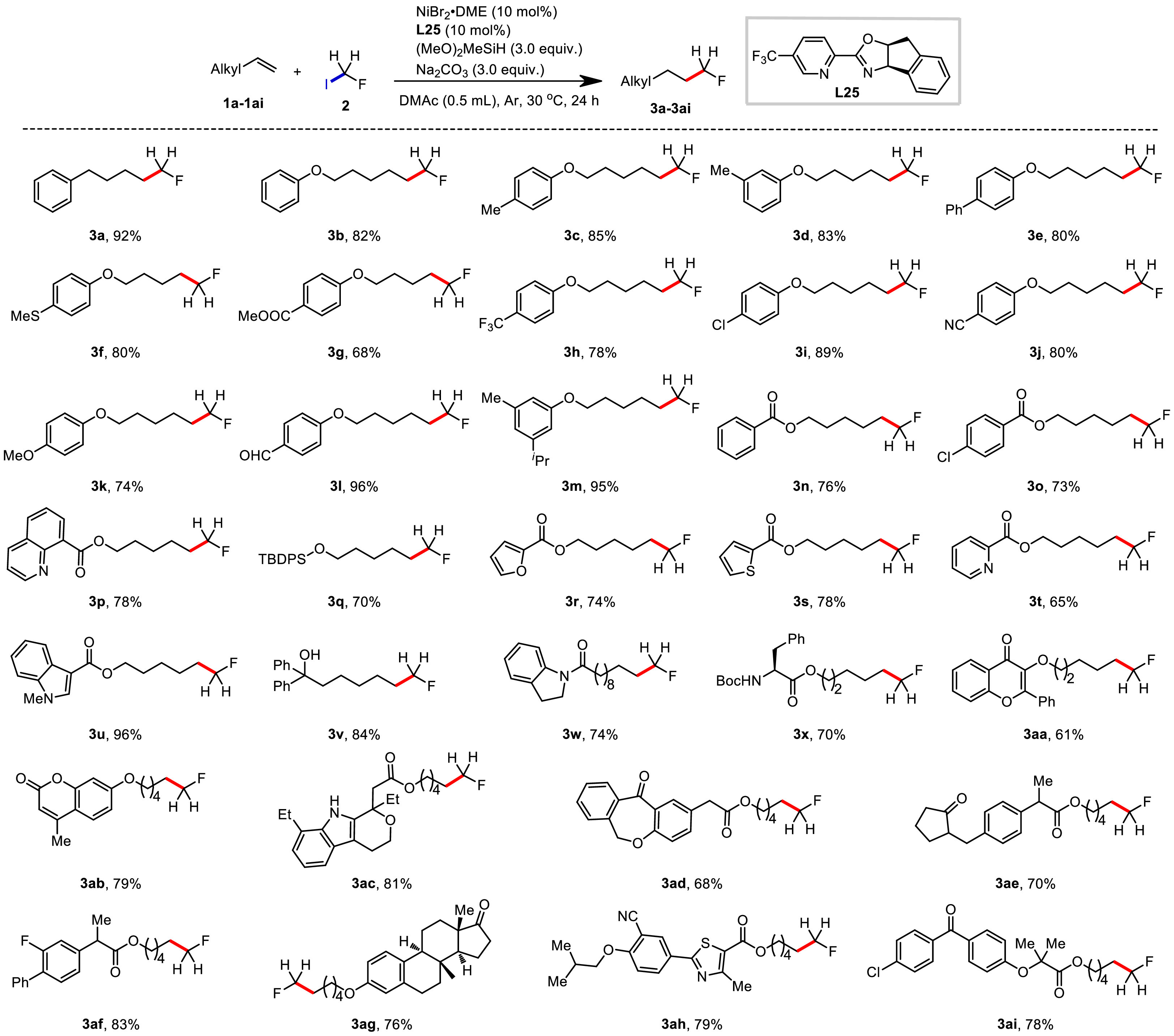
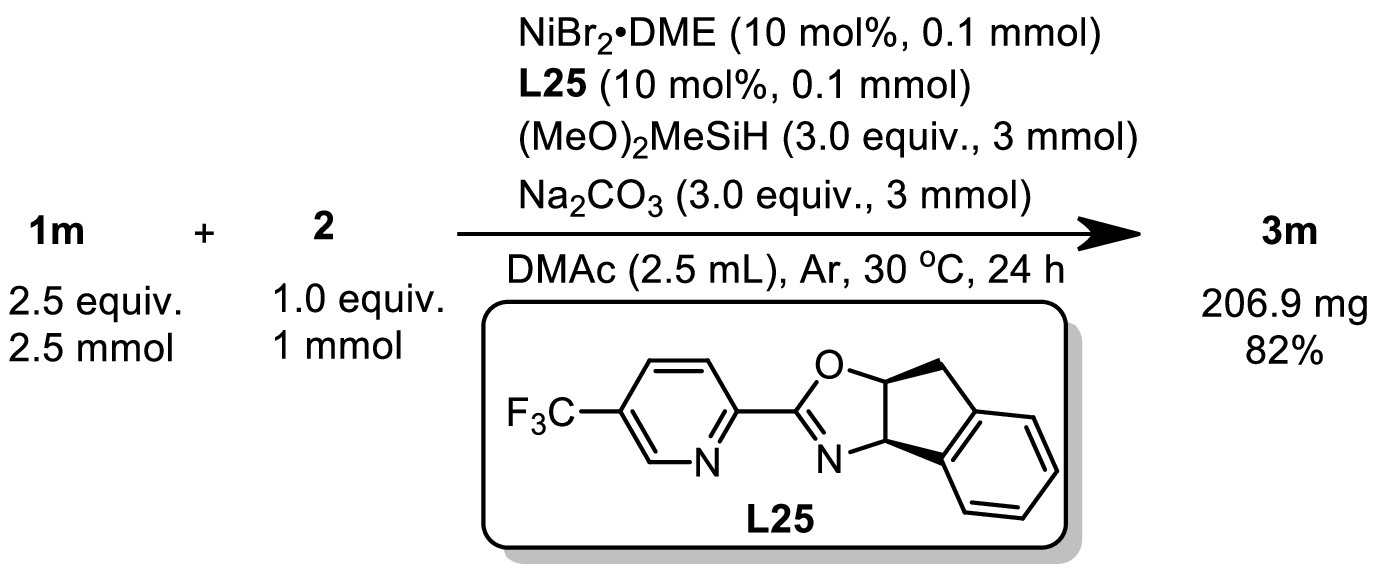
With the optimized conditions in hand, we examined the alkenes scope (Scheme 2). First, alkenes tethered with substituted phenyl ethers were used to evaluate the functional group tolerance. Good to excellent yields (68%–96%) were observed with numerous functional groups, including electron-neutral alkyl (3b-3d, 3m) and aryl (3e) groups, electron-donating groups such as thiomethyl (3f), methoxy (3k) and electron-withdrawing groups such as ester (3g), trifluoromethyl (3h), chloride (3i), cyano (3j) and formyl (3l). Silyl ether (3q) and benzoate derivatives (3n) were also well tolerated, giving 70%–76% yields. Of note we were able to prepare 3v, which contains a free hydroxyl group, and 3x, an amino acid derivative, in 84% and 70% yields respectively. Considering the importance of heterocycles as building blocks in medicinal chemistry, we next screened various representative heterocyclic compounds. We were pleased that coordinating atoms like nitrogen, oxygen and sulfur, which typically pose challenges in transition metal catalysis, were all compatible under our reaction conditions. For example, this transformation was effective with quinoline (3p), furan (3r), thiophene (3s), pyridine (3t), N-methyl indole (3u) and amide (3w), affording the desired products in 65%−96% yields. After demonstrating the excellent functional group tolerance and substrate compatibility, the synthetic potential of this method was then elucidated via monofluoromethylation of drug candidates. Alkene species bearing bioactive motifs such as flavonols (3aa), 4-methylumbelliferone (3ab), etodolac (3ac), isoxepac (3ad), loxoprofen (3ae), flurbiprofen (3af), estrone (3ag), febuxostat (3ah) and fenofibric acid (3ai) could be smoothly monofluoromethylated with gratifying yields (61%–83%), showcasing its great potential to be an efficient and expedient tactic for drug discovery. This method could be easily scaled up and allowed for isolation of 82% yield, indicating that it could be used for industrial-grade synthesis process (Scheme 3).

|
Download:
|
| Scheme 2. Scope of unactivated alkenes. Reaction conditions: 1 (0.5 mmol, 2.5 equiv.), 2 (0.2 mmol, 1.0 equiv.), NiBr2·DME (10 mol%), L25 (10 mol%), (MeO)2MeSiH (3.0 equiv.), Na2CO3 (3.0 equiv.), DMAc (0.5 mL), Ar, 30 ℃, 24 h. Isolated yield. | |
{kind=link}

|
Download:
|
| Scheme 3. Mmol scale experiment. | |
{kind=link}
In analogy to earlier work, we surmised that the reaction may proceed via a radical pathway and a series of control experiments were carried out to verify this proposal (Scheme 4). First, 2.0 equiv. of free radical inhibitor TEMPO was added to the standard reaction system and found no product, while product 4 was detected by HRMS (Scheme 4a), indicating that there contains a monofluoromethyl radical in this process. Meanwhile, (2-vinylcyclopropyl)benzene 5 was then subjected to the standard conditions to verify the reaction sequence of Ni-H species and radical species with alkenes and the product 6 was obtained in 30% yield, while the ring-opening product normally furnished by fluoromethyl radical attack first to alkene was not isolated (Scheme 4b), which indicated that the concentration of free radicals was very low. This result indicates that the reation was initiated by the insertion of Ni-H species to the alkenes rather than the addition of the monofluoromethyl radical to the alkenes.

|
Download:
|
| S4. Mechanistic studies. | |
{kind=link}
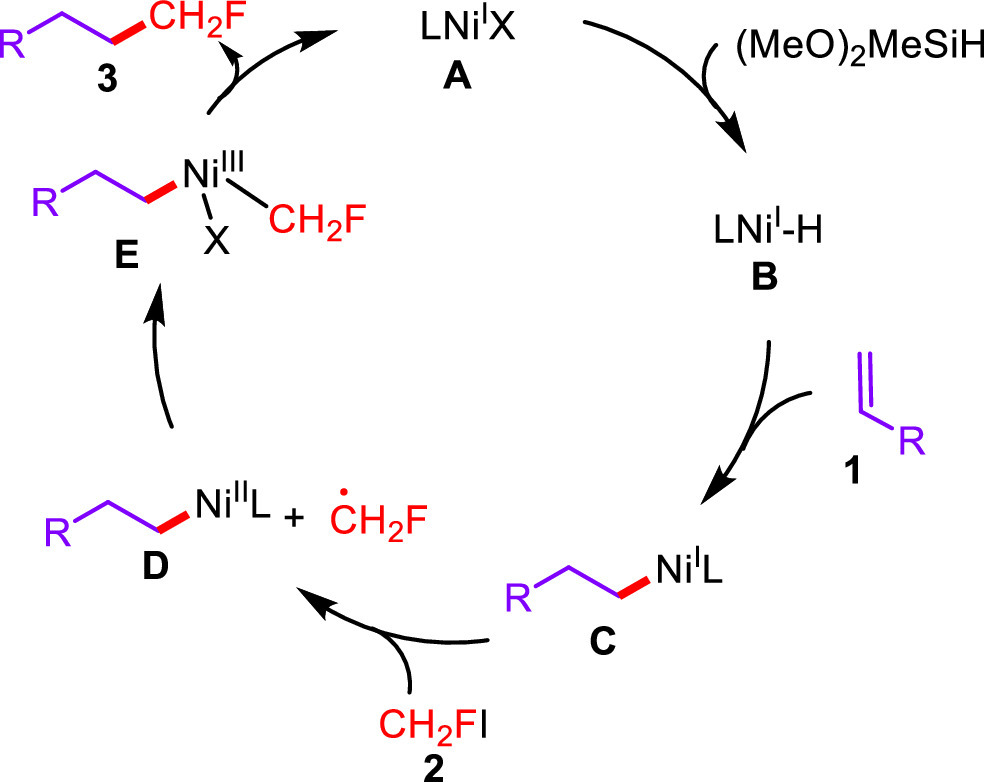
Based on our observations and previous reports [45-49], the plausible mechanism involving a NiI/NiIII catalytic cycle was depicted in Scheme 5. With the presence of Na2CO3, the hydrogen atom transfer between NiI species A and silane generated Ni-H species B, which offered alkyl-nickel intermediate C after the following insertion by alkene. Single electron oxidation of NiI species C by fluoroiodomethane 2 afforded monofluoromethyl radical and NiII intermediate D, and radical oxidation of D furnished NiIII species E. Finally, the reductive elimination from E furnished monofluoroalkane 3 and regenerated NiI catalyst A.

|
Download:
|
| Scheme 5. Proposed mechanism. | |
{kind=link}
In conclusion, we have developed a nickel-catalyzed three-component hydrofluoromethylation of unactivated alkenes with industrial raw material CH2FI. This method enables the use of readily available and low-cost alkenes, thereby complementing the existing reductive cross-coupling reactions which requires the preparation of its alkyl halide counterparts. The transformation tolerates a diverse collection of functional groups and bioactive compounds. While this strategy is expected to flourish in the coming years with growing applications in the drug discovery, further exploration of the detailed mechanism and more synthetic applications was still ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentWe gratefully acknowledge the National Natural Science Foundation of China (No. 21971228) for financial support.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.06.037.
| [1] |
C. Ni, J. Hu, Chem. Soc. Rev. 45 (2016) 5441-5454. DOI:10.1039/C6CS00351F |
| [2] |
R. Berger, G. Resnati, P. Metrangolo, et al., Chem. Soc. Rev. 40 (2011) 3496-3508. DOI:10.1039/c0cs00221f |
| [3] |
M.C. Walker, M.C.Y. Chang, Chem. Soc. Rev. 43 (2014) 6527-6536. DOI:10.1039/C4CS00027G |
| [4] |
J. Wang, M. Sanchez-Rosello, J.L. Acena, et al., Chem. Rev. 114 (2014) 2432-2506. DOI:10.1021/cr4002879 |
| [5] |
N.A.J. Meanwell, Med. Chem. 61 (2018) 5822-5880. DOI:10.1021/acs.jmedchem.7b01788 |
| [6] |
T. Liang, C.N. Neumann, T. Ritter, Angew. Chem. Int. Ed. 52 (2013) 8214-8264. DOI:10.1002/anie.201206566 |
| [7] |
M.G. Campbell, T. Ritter, Chem. Rev. 115 (2015) 612-633. DOI:10.1021/cr500366b |
| [8] |
D.A. Petrone, J. Ye, M. Lautens, Chem. Rev. 116 (2016) 8003-8104. DOI:10.1021/acs.chemrev.6b00089 |
| [9] |
S. Barata-Vallejo, M.V. Cooke, A. Postigo, ACS Catal. 8 (2018) 7287-7307. DOI:10.1021/acscatal.8b02066 |
| [10] |
R. Szpera, D.F.J. Moseley, L.B. Smith, et al., Angew. Chem. Int. Ed. 58 (2019) 14824-14848. DOI:10.1002/anie.201814457 |
| [11] |
M. Reichel, K. Karaghiosoff, Angew. Chem. 132 (2020) 12364-12377. DOI:10.1002/ange.201913175 |
| [12] |
G.S. Lal, G.P. Pez, R.J. Pesaresi, et al., J. Org. Chem. 64 (1999) 7048-7054. DOI:10.1021/jo990566+ |
| [13] |
H. Dang, M. Mailig, G. Lalic, Angew. Chem. Int. Ed. 53 (2014) 6473-6476. DOI:10.1002/anie.201402238 |
| [14] |
J. Guo, C. Kuang, J. Rong, et al., Chem. Eur. J. 25 (2019) 7259-7264. DOI:10.1002/chem.201901176 |
| [15] |
V. Fasano, N. Winter, A. Noble, et al., Angew. Chem. Int. Ed. 59 (2020) 8502-8506. DOI:10.1002/anie.202002246 |
| [16] |
Y. Li, C. Ni, J. Liu, et al., Org. Lett. 8 (2006) 1693-1696. DOI:10.1021/ol060322t |
| [17] |
J. Liu, L. Zhang, J. Hu, Org. Lett. 10 (2008) 5377-5380. DOI:10.1021/ol802226k |
| [18] |
K. Matsuzaki, T. Furukawa, E. Tokunaga, et al., Org. Lett. 15 (2013) 3282-3285. DOI:10.1021/ol4013102 |
| [19] |
Y. Geng, A. Liang, X. Gao, et al., J. Org. Chem. 82 (2017) 8604-8610. DOI:10.1021/acs.joc.7b01438 |
| [20] |
G. Parisi, M. Colella, S. Monticelli, et al., J. Am. Chem. Soc. 139 (2017) 13648-13651. DOI:10.1021/jacs.7b07891 |
| [21] |
G.K.S. Prakash, I. Ledneczki, S. Chacko, et al., Org. Lett. 10 (2008) 557-560. DOI:10.1021/ol702500u |
| [22] |
Y. Zhao, B. Gao, C. Ni, et al., Org. Lett. 14 (2012) 6080-6083. DOI:10.1021/ol3029737 |
| [23] |
H. Doi, I. Ban, A. Nonoyama, et al., Chem. Eur. J. 15 (2009) 4165-4171. DOI:10.1002/chem.200801974 |
| [24] |
N.Y. Wu, X.H. Xu, F.L. Qing, ACS Catal. 9 (2019) 5726-5731. DOI:10.1021/acscatal.9b01530 |
| [25] |
Y. Cao, L. Jiang, W. Yi, Adv. Synth. Catal. 361 (2019) 4360-4368. DOI:10.1002/adsc.201900480 |
| [26] |
X. Shen, M. Zhou, C. Ni, et al., Chem. Sci. 5 (2014) 117-122. DOI:10.1039/C3SC51831K |
| [27] |
Z. He, P. Tan, C. Ni, et al., Org. Lett. 17 (2015) 1838-1841. DOI:10.1021/acs.orglett.5b00308 |
| [28] |
N. Noto, T. Koike, M. Akita, ACS Catal. 9 (2019) 4382-4387. DOI:10.1021/acscatal.9b00473 |
| [29] |
X. Jiang, S. Sakthivel, K. Kulbitski, et al., J. Am. Chem. Soc. 136 (2014) 9548-9551. DOI:10.1021/ja504089y |
| [30] |
X. Jiang, M. Gandelman, J. Am. Chem. Soc. 137 (2015) 2542-2547. DOI:10.1021/jacs.5b00473 |
| [31] |
Y.M. Su, G.S. Feng, Z.Y. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 6003-6007. DOI:10.1002/anie.201412026 |
| [32] |
J. Sheng, H.Q. Ni, H.R. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 7634-7639. DOI:10.1002/anie.201803228 |
| [33] |
J. Sheng, H.Q. Ni, S.X. Ni, et al., Angew. Chem. Int. Ed. 60 (2021) 15020-15027. DOI:10.1002/anie.202102481 |
| [34] |
R. Cu, J. Sheng, B.B. Wu, et al., Chem. Commun. 57 (2021) 9084-9087. DOI:10.1039/D1CC02837E |
| [35] |
Y.L. Li, W. Liu, Z.Y. Liu, CCS Chem. 3 (2021) 3320-3328. |
| [36] |
C.J. DeSantis, S.E. Skrabalak, J. Am. Chem. Soc. 135 (2013) 3989-3996. DOI:10.1021/ja312271c |
| [37] |
X. Wu, F.A. Cruz, A. Lu, et al., J. Am. Chem. Soc. 140 (2018) 10126-10130. DOI:10.1021/jacs.8b06069 |
| [38] |
X. Li, J. Jin, P. Chen, Nat. Chem. 14 (2022) 425-432. DOI:10.1038/s41557-021-00869-x |
| [39] |
M. Arlt, O. Hindsgaul, J. Org. Chem. 60 (1995) 4617-4628. DOI:10.1021/jo00119a045 |
| [40] |
Q. Liu, Y. Lu, H. Sheng, et al., Angew. Chem. Int. Ed. 60 (2021) 25477-25484. DOI:10.1002/anie.202111006 |
| [41] |
Y. Zhang, B. Han, S. Zhu, Angew. Chem. Int. Ed. 58 (2019) 13860-13864. DOI:10.1002/anie.201907185 |
| [42] |
Y. He, C. Liu, L. Yu, et al., Angew. Chem. Int. Ed. 59 (2020) 9186-9191. DOI:10.1002/anie.202001742 |
| [43] |
D. Qian, Sr. Bera, X. Hu, J. Am. Chem. Soc. 143 (2021) 1959-1967. DOI:10.1021/jacs.0c11630 |
| [44] |
Y. Cheng, Z. Gui, R. Tao, et al., Green Synth. Catal. 3 (2022) 377-379. DOI:10.1016/j.gresc.2022.03.009 |
| [45] |
Y. He, Y. Cai, S. Zhu, J. Am. Chem. Soc. 139 (2017) 1061-1064. DOI:10.1021/jacs.6b11962 |
| [46] |
Z.Y. Wang, G.H. Wan, G.Y. Wang, et al., Tetrahedron Lett. 59 (2018) 2302-2305. DOI:10.1016/j.tetlet.2018.05.008 |
| [47] |
Z. Wang, H. Yin, G.C. Fu, Nature 563 (2018) 379-383. DOI:10.1038/s41586-018-0669-y |
| [48] |
S. Wang, J.X. Zhang, T.Y. Zhang, et al., Nat. Commun. 12 (2021) 2771-2780. DOI:10.1038/s41467-021-22983-7 |
| [49] |
W. Guo, L. Cheng, G. Ma, et al., Org. Lett. 24 (2022) 1796-1801. DOI:10.1021/acs.orglett.2c00148 |