 2023, Vol. 34
2023, Vol. 34 


b Fine Chemical Industry Research Institute, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China;
c Huizhou Research Institute, Sun Yat-sen University, Huizhou 516081, China;
d Advanced Energy Science and Technology Guangdong Laboratory, Huizhou 516003, China
As a bulk chemical, propylene oxide (PO) is mainly used as a raw material in the production of polyether polyols and propylene glycol, which are widely used in pharmaceuticals, food, textiles, and other industries [1-3]. Currently, global annual PO production is more than 10 million tons, and demand is increasing annually. At present, the main industrial production methods of PO are chlorohydrin and peroxidation processes, which suffer from problems such as environmental impact, high cost, and security [4]. Thus, the development of efficient strategies to achieve the direct epoxidation of propylene by molecular oxygen with excellent selectivity is highly desirable [5-7].
Industrially, silver has been successfully used in the epoxidation of ethylene with high selectivity (> 90%). However, Ag-based catalysts are not suitable for propylene epoxidation because they result in the oxidation of allylic C—H bonds before C=C bonds [8]. Numerous attempts have been made to modify Ag-based catalysts to improve selectivity for PO, but their efficiencies are not high enough for industrial-scale applications [9-11]. Other gas-phase catalytic systems using Au [12-14], Mo [15-17], Cu [18-20], and the perovskite LaCoO3 as the catalyst [21] have been developed for the direct epoxidation of propylene. However, due to the thermodynamic limitations of the propylene molecule, the PO selectivity of these systems is still unsatisfactory under high-temperature conditions.
The epoxidation of propylene in homogeneous catalytic systems is an alternative strategy for improving selectivity of PO under mild conditions [22-24]. Metalloporphyrins are widely used as catalyst in the oxidation of hydrocarbons or oxygen electrocatalysis [25-32]. However, there are few existing examples of the direct metalloporphyrin-catalyzed epoxidation of propylene in homogeneous systems. A manganese porphyrin-mediated liquid-phase epoxidation of propylene with molecular oxygen and benzaldehyde as the co-substrate was developed by our group [33]. In addition, iron porphyrins have shown excellent PO selectivity in direct propylene epoxidation by using acrolein as a co-substrate at a 1:1 nacrolein/npropylene molar ratio [34]. More recently, we developed a direct propylene epoxidation protocol by using ruthenium porphyrin and cerium(IV) sulfate as cocatalysts, achieving a PO selectivity of 82.3% [35].
Isoprene is a bulk chemical mainly used as a raw material in the petrochemical industry to synthesize rubber and butyl rubber monomer [36]. Isoprene is a conjugated diene with an allyl methyl group [37]. The α-H of this group can easily be removed to generate an active radical and achieve the activation of molecular oxygen [38-40]. In addition, the products obtained from isoprene oxidation are high value-added chemicals. Therefore, using isoprene as a co-substrate to active molecular oxygen is expected to realize the direct and simultaneous epoxidation of propylene and isoprene.
With this strategy in mind, a protocol on the direct epoxidation of propylene catalyzed by metalloporphyrins in the presence of isoprene has been developed and is reported in this work. The influence of various reaction parameters such as the catalyst, solvent, reaction temperature, and oxygen pressure on the activity and selectivity toward PO was evaluated. A high-valence Mn-oxo species was confirmed by mass spectroscopy and operando UV–vis spectroscopy. The role of isoprene was demonstrated, and a plausible mechanism was proposed. This proposed protocol provides a possible approach for simultaneously manufacturing propylene oxide and isoprene monoxide in the petrochemical industry.
In a blank experiment, only 7% of the propylene was converted to corresponding products in the presence of isoprene (Table 1, entry 1). When the reaction was mediated by metal salts like Co(OAc)2 or MnCl2, the yields of PO were unsatisfactory (Table 1, entries 2–3). Compared with the blank experiment and metal salts, the addition of the metalloporphyrin catalyst (0.01 mol%, based on propylene) resulted in an increased the propylene conversion and PO selectivity (Table 1, entries 4–8). Because cobalt, ruthenium, iron, and manganese porphyrins (the structures as shown in Fig. S1 in Supporting information) have been reported as effective homogeneous catalysts for epoxidation with molecular oxygen, the catalytic performance of these catalysts was examined. Manganese porphyrins were preferred for propylene epoxidation in the presence of isoprene. The superior performance of the Mn complexes can be ascribed to their efficient charge transfer properties. This results in a superior oxygen rebound, leading to enhanced activity and PO selectivity. Compared with MnTPPCl, Mn(p–OCH3)TPPCl exhibited better catalytic performance and PO selectivity (Table 1, entries 7 and 8). This can be attributed to the ligand T(p–OCH3)PP efficiently increasing the charge density around the central metal manganese [41-43].
|
|
Table 1 Epoxidation of propylene catalyzed by metalloporphyrins with isoprene.a |
Subsequently, it was determined that the conversion of propylene increased as the catalyst dose increased (Table 1, entries 9 and 10). However, no significant difference in efficiency was observed when the amount of catalyst was higher than 0.1 mol% (based on propylene). It could be attributed to the self-polymerization of metalloporphyrins catalyst [44]. It was also determined that the selectivity of PO was not significantly affected by the amount of catalyst.
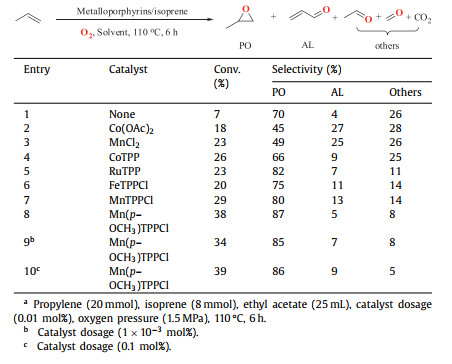
The presence of isoprene allows oxygen to be activated via the active allyl radical generated from the abstraction of α-hydrogen. Therefore, the effect of isoprene concentration on propylene epoxidation was examined (Fig. 1). In a control experiment without isoprene, the conversion of propylene was nearly 5%. This is indicative that isoprene is crucial for oxygen activation in the epoxidation mechanism. A higher isoprene/propylene molar ratio improved propylene conversion, which increased to 38% at nisoprene/npropylene = 0.4. However, when the amount of isoprene was increased beyond this, no significant efficiency improvements were observed. The increase in isoprene concentration had little effect on the selectivity of the main oxidized products. In our previous work with benzaldehyde as co-substrate, the amount of 38% conversion of propylene and 80% PO selectivity was obtained at nbenzaldehyde/npropylene = 1/1 [33]. In contrast, the amount of co-substrate in this catalytic protocol was reduced significantly.

|
Download:
|
| Fig. 1. Propylene epoxidation at different molar ratios of isoprene/propylene. Propylene (20 mmol), ethyl acetate (25 mL), Mn(p–OCH3)TPPCl (0.01 mol%), oxygen pressure (1.5 MPa), 110 ℃, 6 h. | |
{kind=link}
As a co-substrate, the oxidation of isoprene was also investigated. The only oxidized product was isoprene monoxide (2-methyl-2-vinyloxirane). With an increase in isoprene concentration, the conversion of isoprene gradually decreased. This indicates that autooxidation did not occur in this catalytic system. This was further confirmed by the consumption of isoprene during propylene epoxidation. When the molar ratio of isoprene/propylene was 0.4 (the conversion of propylene was 38%), the consumption of isoprene was almost the same as that of propylene. Even when the isoprene/propylene molar ratio was increased to 1:1, the consumption of isoprene did not significantly change. Therefore, it can be concluded that polymerization of isoprene does not easily occur under the reaction conditions. This conclusion was confirmed by GPC (gel permeation chromatography) characterization of the reaction solution.
In the liquid phase epoxidation of propylene, the solvent plays a key role by affecting the solubility of propylene and via the solvation effect. As shown in Table S1 (Supporting information), the conversion of propylene was not significantly affected by solvent polarity. A propylene conversion of higher than 25% was achieved in both the polar and nonpolar solvents. Propylene has excellent solubility in methanol. Hence, methanol was favorable for propylene conversion, but not for the generation of PO (Table S1, entry 2) in this catalytic system. In contrast, high propylene conversion and better selectivity toward PO were obtained in ethyl acetate (Table S1, entry 3).
Conversion and selectivity are closely related to reaction temperature. As presented in Fig. S2 (Supporting information), the conversion of propylene was significantly influenced by the reaction temperature. With increasing temperature, propylene conversion also increased. Furthermore, increasing the reaction temperature from 70 ℃ to 120 ℃ substantially increased the reaction rate, with conversion significantly improving from 4% to 42%. This indicates that higher temperatures promote the formation of free radicals in the catalytic system, increasing the reaction rate. However, the selectivity toward PO decreased under higher temperatures, especially when the reaction temperature was higher than 110 ℃. Meanwhile, the selectivity toward acetaldehyde increased at high temperatures.
The effect of oxygen pressure on propylene epoxidation was also examined (Fig. S3 in Supporting information). Propylene conversion generally increased with increasing oxygen pressure, but the rate of increase was not significant when the oxygen pressure was higher than 1.5 MPa. It was also determined that the oxygen pressure had little influence on the selectivity of products. From the viewpoints of mild conditions and efficiency, the oxygen pressure should be kept below 2.0 MPa.
A similar trend was seen for the conversion of isoprene. Increasing the oxygen pressure from 0.5 MPa to 1.0 MPa enhanced the conversion of isoprene from 85% to 98%. No significant changes in isoprene conversion were observed as the oxygen pressure increased above 1.0 MPa. The high selectivity of isoprene monoxide did not change with increasing oxygen pressure. This indicates that neither autooxidation nor polymerization occur under the examined conditions.
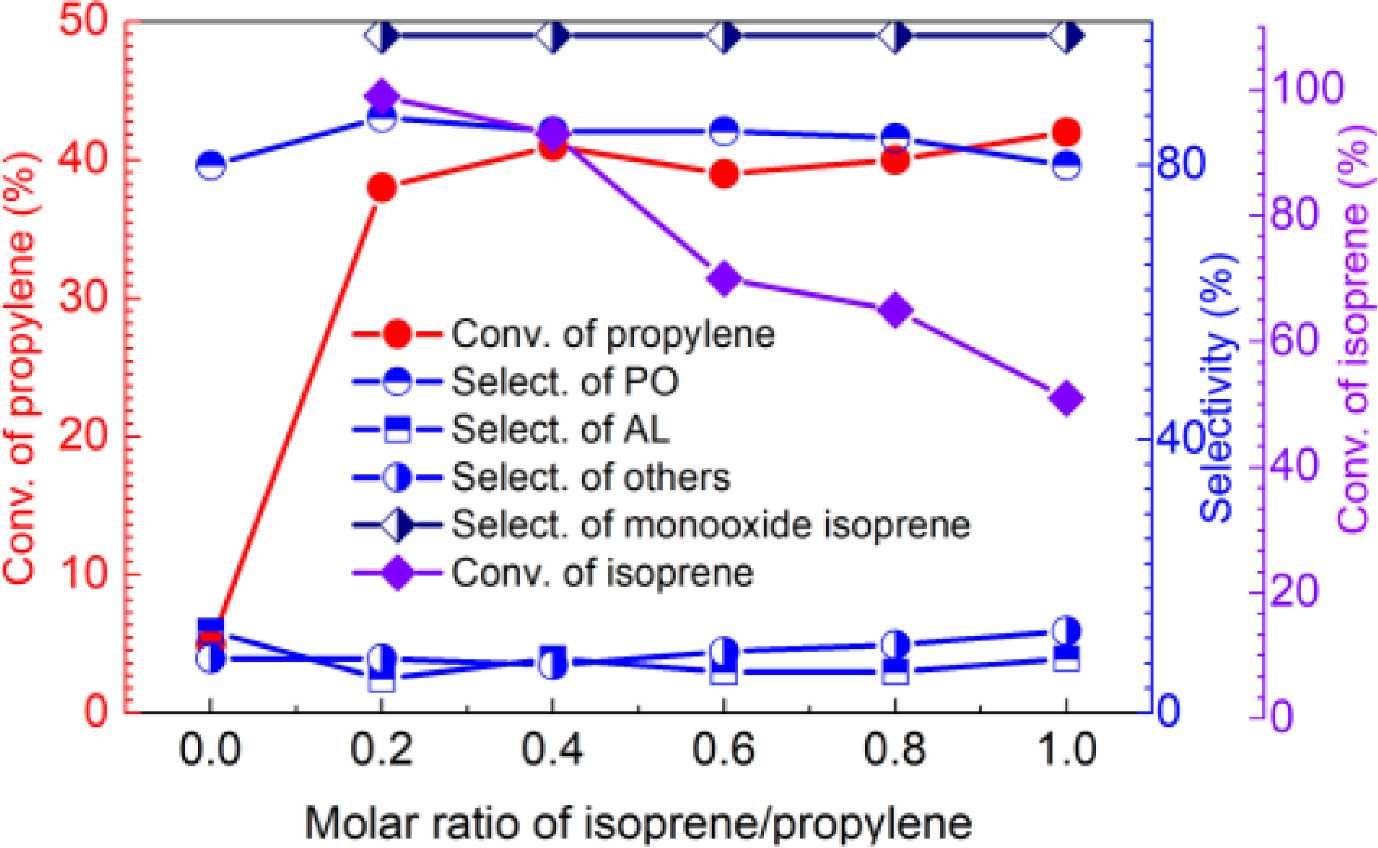
The profile of propylene epoxidation catalyzed by Mn(p–OCH3)TPPCl in the presence of isoprene and molecular oxygen is shown in Fig. 2a. As can be seen, there was an induction period of nearly 2 h. After this, the reaction rate rapidly accelerated, demonstrating a typical free radical reaction profile. A similar reaction profile was obtained for isoprene oxidation with the same 2 h induction period (Fig. 2b). This indicates that the initiation of free radicals is the rate-limiting step [35].

|
Download:
|
| Fig. 2. The reaction profiles of propylene (a) and isoprene (b) epoxidation catalyzed by MnTPPCl. Propylene (20 mmol), isoprene (8 mmol), ethyl acetate (25 mL), Mn(p–OCH3)TPPCl (0.01 mol%), oxygen pressure (1.5 MPa), 110 ℃. | |
{kind=link}
In addition, the selectivity toward the main propylene oxidation products did not significantly change during the reaction process. This also indicates that the three main products are generated by different parallel reaction pathways. Furthermore, as shown in Fig. 2b, no products other than isoprene monoxide were observed in isoprene oxidation.
As shown in Fig. 2, the epoxidation of propylene with isoprene exhibited the features of a radical-involved reaction. To investigate the free radical mechanism, a free radical inhibitor (2, 6-di‑tert-butylphenol (BTH), 0.5 mmol) was added to the solution. It was observed that propylene epoxidation was subsequently quenched. Furthermore, the conversion of isoprene to isoprene monoxide was also terminated (Fig. S4 in Supporting information).
Using MnTPPCl as the catalyst, the epoxidation of propylene was conducted in a stainless-steel reactor connected to an AvaSpec-2048 spectrometer to obtain operando UV–vis spectra of the reaction solution. The spectrophotometer was programmed to acquire UV–vis spectra at an interval of 6 min (Fig. 3).

|
Download:
|
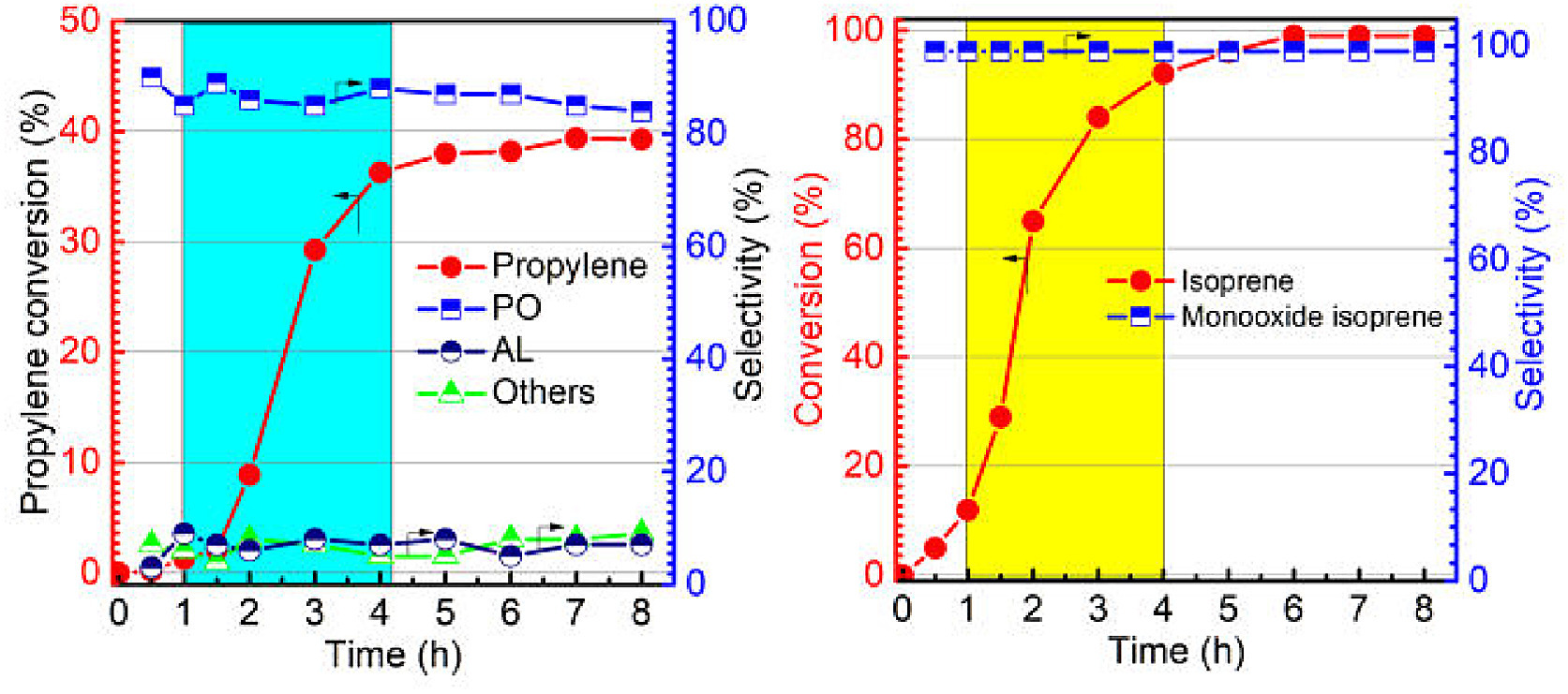
| Fig. 3. Operando UV–vis spectra of propylene epoxidation by manganese porphyrin with isoprene and molecular oxygen. Reaction conditions: Propylene (20 mmol), isoprene (8 mmol), ethyl acetate (25 mL), MnTPPCl (0.01 mol%), oxygen pressure (1.5 MPa), 110 ℃, spectra measured at intervals of 6 min. Insert: absorbance trend of characteristic peaks with time. | |
{kind=link}
Fig. 3 shows that a characteristic inverted peak at 299 nm was observed after the addition of isoprene. As the reaction proceeded, the inverted peak at 299 nm slowly declined in intensity due to the induction period of the free radical reaction. After 90 min, a red shift of the characteristic peak from 299 nm to 304 nm was observed. The intensity of the peak at 304 nm then rapidly increased. The peak at 304 nm can be ascribed to the typical absorption of isoprene. This change in absorption suggests that the conjugation effect of isoprene was enhanced. Furthermore, the time of the change in absorption coincided with the reaction profiles (Fig. 2), reflecting the process of free radical initiation.
The absorbance peaks at 470 nm and 611 nm are the characteristic Soret and Q band absorption of the MnTPPCl catalyst, respectively [45]. At the initial stage of reaction, no changes in the Soret and Q band absorption peaks were observed. After about 3 h, the Q band at 612 nm gradually decreased in intensity and the formation of a new intermediate with an electronic absorption band at 421 nm was observed. The peak at 421 nm can be ascribed to the characteristic absorption of the Mn-oxo high-valence species PorMnIV=O [33, 46, 47]. The intensity of the absorption peak at 421 nm initially increased and then stabilized, ensuring that the catalytic cycle produced oxidized products (Fig. 3, inset). In addition, a MALDI-TOF-MS (matrix-assisted laser desorption ionization time-of-flight mass spectrometry) spectrum of the resulting solution exhibited a prominent ion peak at an m/z of 682.430. The corresponding mass and isotope distribution patterns can be attributed to the high-valence species TPPMnIV=O (Fig. S5 in Supporting information). This suggested that the new species may be the Mn-oxo species.
Operando infrared spectroscopy was employed to monitor the course of propylene epoxidation. First, in order to further investigate the reaction process, an operando infrared spectrometer was used to track the reaction (Fig. S6 in Supporting information). The IR characteristic peaks of isoprene and solvent acetonitrile were observed (Fig. S6). The change of isoprene in solution was characterized by tracking the characteristic peak at 993 cm−1 [48]. For propylene, the characteristic stretching vibration absorption of the C=C bond at 1647 cm−1 and the allyl C—H peak at 913 cm−1 were monitored [49].
From the operando IR spectra, it was determined that the intensity of propylene peaks at 913 cm−1 and 1647 cm−1 and the isoprene peak at 993 cm−1 rapidly declined at the beginning of signal recording (Fig. S7 in Supporting information). This can be ascribed to the dissolving process of propylene and isoprene in the solvent. Then, as the reaction proceeded, the intensity of the characteristic peaks steadily declined. The intensity of peaks at 1740 cm−1 and 1148 cm−1 rapidly increased when the reaction time was longer than 4 h. These peaks are ascribed to the characteristic stretching vibration of the epoxy groups of isoprene monoxide and PO. The intensity trends of the operando IR spectra were consistent with those of the UV–vis spectra and the reaction profiles.
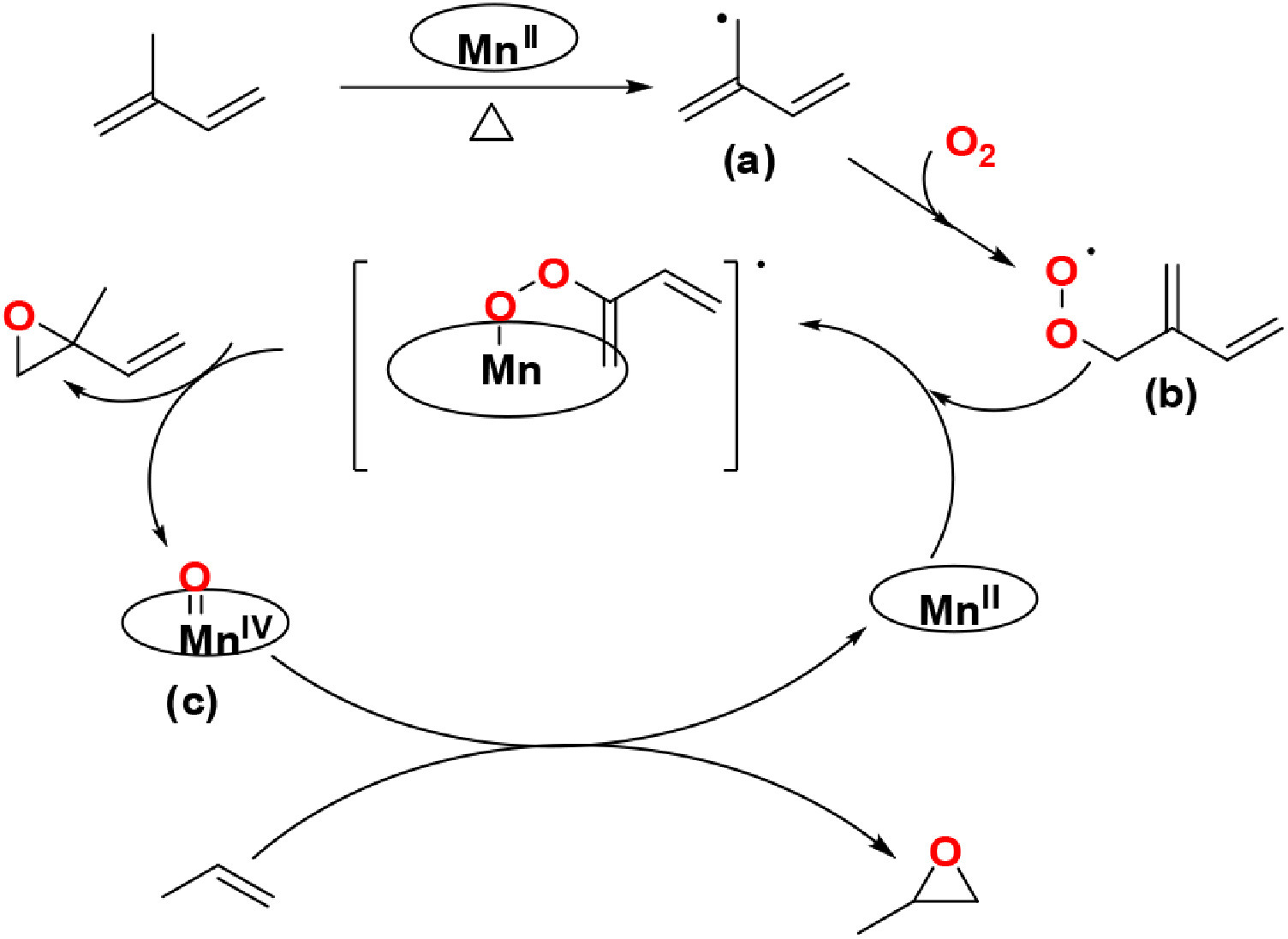
Based on these experiments, a plausible mechanism for propylene epoxidation catalyzed by manganese porphyrins in the presence of isoprene and molecular oxygen can be proposed, as shown in Fig. 4. The α-hydrogen atom of isoprene′s allyl group is easily abstracted at high temperatures to generate an allylic radical (a). An allyl peroxyl radical (b) is rapidly formed by the combination of one oxygen molecule with the allylic radical (a). Subsequently, the allyl peroxyl radical (b) reacts with the manganese porphyrins, generating isoprene monoxide and the high-valent Mn porphyrin intermediate (c). The formation of PO can then be attributed to oxygen rebound through the high-valent Mn species.

|
Download:
|
| Fig. 4. Plausible mechanism of propylene epoxidation catalyzed by manganese porphyrins in the presence of isoprene and molecular oxygen. | |
{kind=link}
In summary, a protocol for the simultaneous preparation of propylene oxide and isoprene monoxide was developed. The manganese porphyrin Mn(p–OCH3)TPPCl showed high efficiency for the direct liquid phase catalyzed epoxidation of propylene in the presence of isoprene. In this reaction system, the conversion of propylene was 38% and the selectivity toward PO was 87%. Moreover, a high-valence Mn-oxo species was confirmed by mass spectroscopy and operando UV–vis spectroscopy. The role of isoprene was determined, and a plausible reaction mechanism was proposed.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Key Research and Development Program of China (No. 2020YFA0210900), the National Natural Science Foundation of China (Nos. 21938001 and 21878344), Guangdong Provincial Key R & D Programme (No. 2019B110206002), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No. 2017BT01C102).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.07.001.
| [1] |
L. Cheng, C.R. Yin, F. Mehmood, et al., ACS Catal. 4 (2014) 32-39. DOI:10.1021/cs4009368 |
| [2] |
A. Marimuthu, J.W. Zhang, S. Linic, Science 339 (2013) 1590-1593. DOI:10.1126/science.1231631 |
| [3] |
D.H. Wells, W.N. Delgass, K.T. Thomson, J. Am. Chem. Soc. 126 (2004) 2956-2962. DOI:10.1021/ja037741v |
| [4] |
C. Zhan, Q.X. Wang, L.Y. Zhou, et al., J. Am. Chem. Soc. 142 (2020) 14134-14141. DOI:10.1021/jacs.0c03882 |
| [5] |
X.F. Yang, S. Kattel, K. Xiong, et al., Angew. Chem. Int. Ed. 54 (2015) 11946-11951. DOI:10.1002/anie.201504538 |
| [6] |
W.G. Su, S.G. Wang, P.L. Ying, Z.C. Feng, C. Li, J. Catal. 268 (2009) 165-174. DOI:10.1016/j.jcat.2009.09.017 |
| [7] |
A. Seubsai, M. Kahn, S. Senkan, ChemCatChem 3 (2011) 174-179. DOI:10.1002/cctc.201000248 |
| [8] |
T. Baidya, T. Mazumder, K.Y. Koltunov, et al., J. Phys. Chem. C 124 (2020) 14131-14146. DOI:10.1021/acs.jpcc.0c00777 |
| [9] |
I.D. Charisteidis, K.S. Triantafyllidis, Catal. Today 355 (2020) 654-664. DOI:10.1016/j.cattod.2019.06.057 |
| [10] |
Y. Lei, F. Mehmood, S. Lee, et al., Science 328 (2010) 224-228. DOI:10.1126/science.1185200 |
| [11] |
Q. Zhang, G.T. Chai, Y.L. Guo, et al., J. Mol. Catal. A 424 (2016) 65-76. DOI:10.1016/j.molcata.2016.08.019 |
| [12] |
Z.H. Zhang, X. Zhao, G. Wang, et al., AIChE J. 66 (2020) e16815. |
| [13] |
Y.G. Ren, J.H. Huang, Q. Lv, et al., Appl. Catal. A 584 (2019) 117172. DOI:10.1016/j.apcata.2019.117172 |
| [14] |
S.N. Yao, L.H. Xu, J. Wang, et al., Mol. Catal. 448 (2018) 144-152. DOI:10.1016/j.mcat.2018.01.039 |
| [15] |
X.L. Ni, J. Liu, Y.Y. Liu, et al., Chin. Chem. Lett. 28 (2017) 1057-1061. DOI:10.1016/j.cclet.2017.01.020 |
| [16] |
Y.J. Pang, X.H. Chen, C.Z. Xu, Y.J. Lei, K.M. Wei, ChemCatChem 6 (2014) 876-884. DOI:10.1002/cctc.201300811 |
| [17] |
K. Shen, X.H. Liu, G.Z. Lu, et al., J. Mol. Catal. A 373 (2013) 78-84. DOI:10.1016/j.molcata.2013.02.020 |
| [18] |
Q.X. Wang, C. Zhan, L.Y. Zhou, G. Fu, Z.X. Xie, Catal. Commun. 135 (2020) 105897. DOI:10.1016/j.catcom.2019.105897 |
| [19] |
W.M. Zhu, Q.H. Zhang, Y. Wang, J. Phys. Chem. C 112 (2008) 7731-7734. DOI:10.1021/jp800927y |
| [20] |
H. Chu, L. Yang, Q.H. Zhang, Y. Wang, J. Catal. 241 (2006) 225-228. DOI:10.1016/j.jcat.2006.04.028 |
| [21] |
J.Y. Lei, J.J. Dai, K.B. Tan, et al., ACS Sustain. Chem. Eng. 9 (2021) 794-808. DOI:10.1021/acssuschemeng.0c07315 |
| [22] |
Z.W. Xi, N. Zhou, Y. Sun, K.L. Li, Science 292 (2001) 1139-1141. DOI:10.1126/science.292.5519.1139 |
| [23] |
Y.Y. Liu, K. Murata, M. Inaba, Green Chem. 6 (2004) 510-515. DOI:10.1039/b407290c |
| [24] |
N. Zhou, Z.W. Xi, G.Y. Cao, S. Gao, Appl. Catal. A 250 (2003) 239-245. DOI:10.1016/S0926-860X(03)00310-7 |
| [25] |
Y. Morimoto, Y. Shimaoka, Y. Ishimizu, H. Fujii, S. Itoh, Angew. Chem. Int. Ed. 58 (2019) 10863-10866. DOI:10.1002/anie.201901608 |
| [26] |
M. Torrent-Sucarrat, I. Arrastia, A. Arrieta, F.P. Cossio, ACS Catal. 8 (2018) 11140-11153. DOI:10.1021/acscatal.8b01492 |
| [27] |
G. Huang, L.Q. Mo, J.L. Cai, et al., Appl. Catal. B 162 (2015) 364-371. DOI:10.1016/j.apcatb.2014.07.020 |
| [28] |
L.S. Xie, X.P. Zhang, B. Zhao, et al., Angew. Chem. Int. Ed. 60 (2021) 7576-7581. DOI:10.1002/anie.202015478 |
| [29] |
X.L. Li, X.P. Zhang, M. Guo, et al., J. Am. Chem. Soc. 143 (2021) 14613-14621. DOI:10.1021/jacs.1c05204 |
| [30] |
X.L. Li, H.T. Lei, J.Y. Liu, et al., Angew. Chem. Int. Ed. 57 (2018) 15070-15075. DOI:10.1002/anie.201807996 |
| [31] |
L.S. Xie, X.L. Li, B. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 18883-18887. DOI:10.1002/anie.201911441 |
| [32] |
H.Y. Lv, H.B. Guo, K. Guo, et al., Chin. Chem. Lett. 32 (2021) 2841-2845. DOI:10.1016/j.cclet.2021.02.032 |
| [33] |
Y. Li, X.T. Zhou, S.Y. Chen, et al., RSC Adv. 5 (2015) 30014-30020. DOI:10.1039/C4RA15601C |
| [34] |
L.S. Qi, T. Wang, Y.M. Wei, H.S. Tian, Eur. J. Org. Chem. 2018 (2018) 6557-6565. DOI:10.1002/ejoc.201801233 |
| [35] |
X.T. Zhou, H.Y. Yu, Y. Li, C. Xue, H.B. Ji, Ind. Eng. Chem. Res. 59 (2020) 19982-19988. DOI:10.1021/acs.iecr.0c04264 |
| [36] |
P.O. Wennberg, K.H. Bates, J.D. Crounse, et al., Chem. Rev. 118 (2018) 3337-3390. DOI:10.1021/acs.chemrev.7b00439 |
| [37] |
J.R. Monnier, K.T. Peters, G.W. Hartley, J. Catal. 225 (2004) 374-380. DOI:10.1016/j.jcat.2004.04.032 |
| [38] |
X.H. Liu, H.Y. Yu, C. Xue, X.T. Zhou, H.B. Ji, Chin. J. Chem. 38 (2020) 458-464. DOI:10.1002/cjoc.201900426 |
| [39] |
J. Jiang, J.X. Wang, X.T. Zhou, H.Y. Chen, H.B. Ji, Eur. J. Inorg. Chem. (2018) 2666-2674. |
| [40] |
X.T. Zhou, H.B. Ji, Chem. Eng. J. 156 (2010) 411-417. DOI:10.1016/j.cej.2009.10.066 |
| [41] |
H.M. Shen, X. Wang, L. Ning, et al., Appl. Catal. A 609 (2021) 117904. DOI:10.1016/j.apcata.2020.117904 |
| [42] |
H.M. Shen, B. Qi, M.Y. Hu, et al., Catal. Lett. 150 (2020) 3096-3111. DOI:10.1007/s10562-020-03214-y |
| [43] |
H.Y. Chen, M. Lv, X.T. Zhou, et al., Catal. Commun. 109 (2018) 76-79. DOI:10.1016/j.catcom.2018.02.020 |
| [44] |
Y. Yuan, H.B. Ji, Y.X. Chen, et al., Org. Process Res. Dev. 8 (2004) 418-420. DOI:10.1021/op049974s |
| [45] |
C. Poriel, Y. Ferrand, P. Le Maux, J. Rault-Berthelot, G. Simonneaux, Tetrahedron Lett. 44 (2003) 1759-1761. DOI:10.1016/S0040-4039(03)00107-2 |
| [46] |
W.J. Song, M.S. Seo, S.D. George, et al., J. Am. Chem. Soc. 129 (2007) 1268-1277. DOI:10.1021/ja066460v |
| [47] |
M. Guo, M.S. Seo, Y.M. Lee, S. Fukuzumi, W. Nam, J. Am. Chem. Soc. 141 (2019) 12187-12191. DOI:10.1021/jacs.9b04496 |
| [48] |
W.W. Lei, T.P. Russell, L. Hu, et al., ACS Sustain. Chem. Eng. 5 (2017) 5214-5223. DOI:10.1021/acssuschemeng.7b00574 |
| [49] |
J. Herzberger, K. Niederer, H. Pohlit, et al., Chem. Rev. 116 (2016) 2170-2243. DOI:10.1021/acs.chemrev.5b00441 |