 2023, Vol. 34
2023, Vol. 34 


In recent years, the demands for electric energy are growing day by day with the development of economic society and industrial civilization. The declining of traditional fossil energy sources forces people to turn their attention to renewable clean energy resources [1]. However, the energy output of renewable clean energy resources is intermittent, which is difficult to meet the customer requirements. So the reliable and efficient electrical energy storage systems (EESS) are considered as a key support to solve the problem of unstable new energy power generation [2]. The ideal EESS possess the following characteristics: pollution-free, high reliability, low cost, high safety, long cycle life, high gravimetric energy, high energy efficiency, and high power density [3]. Lithium-ion batteries with higher energy density (150–265 Wh/kg) have become the star performer of EESS [4, 5], which change the communication ways and the power supply for portable equipment [6-8]. But "Lithium dendrites" formed on the surface of negative electrodes can penetrate the diaphragm, causing an electrical short circuit. And the use of highly-flammable organic electrolytes has a huge negative impact on the safety of lithium-ion batteries. Beside, the cost of manufacturing lithium-ion batteries has been rising. Therefore, rechargeable aqueous metal-ion batteries are brought into focus due to their high safety and low cost.
Aqueous zinc ion batteries (AZIBs) stand out from numerous rechargeable aqueous metal-ion batteries due to its various advantages, such as high theoretical capacity (820 mAh/g or 5855 mAh/cm3), low redox potential (–0.762 V vs. standard hydrogen electrode), high stability and abundant zinc resources [9-14]. However, many cathode materials paired with zinc anode face more problems including low specific capacity, low lifespan and high solubility [15-17], which can greatly affect the overall electrochemical performances of AZIBs. The commercial success of alkaline zinc-manganese dioxide batteries has led to the rapid development of rechargeable Zn-MnO2 batteries [18-21]. However, the volume expansion and poor conductivity of MnO2, and the formation of irreversible electrochemical-inert products during the charge/discharge process reduce the capacity of Zn-MnO2 batteries and shorten its lifespan [22-25]. Whereafter a variety of cathode materials for AZIBs including vanadium-based derivatives [26-28], Prussian blue analogs (PBAs) [29], and organic compounds [30] are gradually developed. There are few studies on PBAs materials due to their limited capacity (< 150 mAh/g), the oxygen evolution reaction under high operating voltage [31-33] and organic compounds owing to the dissolution in the aqueous electrolyte. Vanadium-based compounds have the characteristics of multiple oxidation states, variable crystal structures, high theoretical capacity (> 300 mAh/g), and highly abundant element in the Earth's crust, which has become a research hotspot [34-39].
In this review, the latest investigations of Zn-ion storage mechanism based on the crystal structures of vanadium-based cathodes was summarized. The flexible VOx polyhedron unit in vanadium-based compounds can be assembled into various open structures such as 1D tunnel, 2D layer and 3D framework by means of different connections to allow for the reversible de/insertion of zinc ions. Additionally, the cations in vanadates as "pillars" can optimize the layer distance to improve the electrochemical performances of AZIBs. The inductive effect of [PO4] tetrahedron in alkali vanadium phosphates can also provide a higher operating voltage for Zn2+ storage. Such variable crystal structures endow vanadium-based compounds excellent electrochemical performance. However, most V-compounds face some problems in inherently poor electrical conductivity, poor structure stability and vanadium dissolution. Finally, the modification strategies for vanadium dissolution and structural instability are proposed for high-performance AZIBs.
2. Vanadium-based materialsVanadium-based materials are very competitive candidates for AZIBs cathodes. Variable ion diffusion channels of vanadium-based materials affect the electrochemical activity of active materials. At present, there are various types of energy storage mechanisms for AZIBs including Zn2+ insertion, H+ insertion, Zn2+/H+ co-insertion, Zn2+/H2O co-insertion and chemical conversion reactions. In this section, the vanadium-based materials are classified based on the material types and their storage mechanisms are discussed based on crystal structures.
2.1. Introduction of vanadium oxides in AZIBsVanadium has 5 valence electrons that can be lost, so it has the ability to adopt multi-oxidation states (from +2 to +5) and to form numerous complexes. The vanadium-oxygen coordination polyhedron in vanadium oxides varies with their oxidation states from tetrahedron through trigonal bipyramid and square pyramid to distorted octahedra and rectilinear octahedra (Fig. 1a), in which the oxidation state of vanadium atom decreases from +5 through +5/+4 to +3 valence [40]. These polyhedrons link to form the chain structure by sharing corner, which link in turn into layers and then three-dimensional (3D) frameworks (Fig. 1b). These open structures can provide a variety of channels for Zn2+ insertion/extraction [41, 42].

|
Download:
|
| Fig. 1. (a) Coordination polyhedrons of vanadium with different valences. Reproduced with permission [40]. Copyright 1999, International Union of Crystallography. (b) The vanadium coordination polyhedron from chains → layers → 3D frameworks by sharing vertices or edges. | |
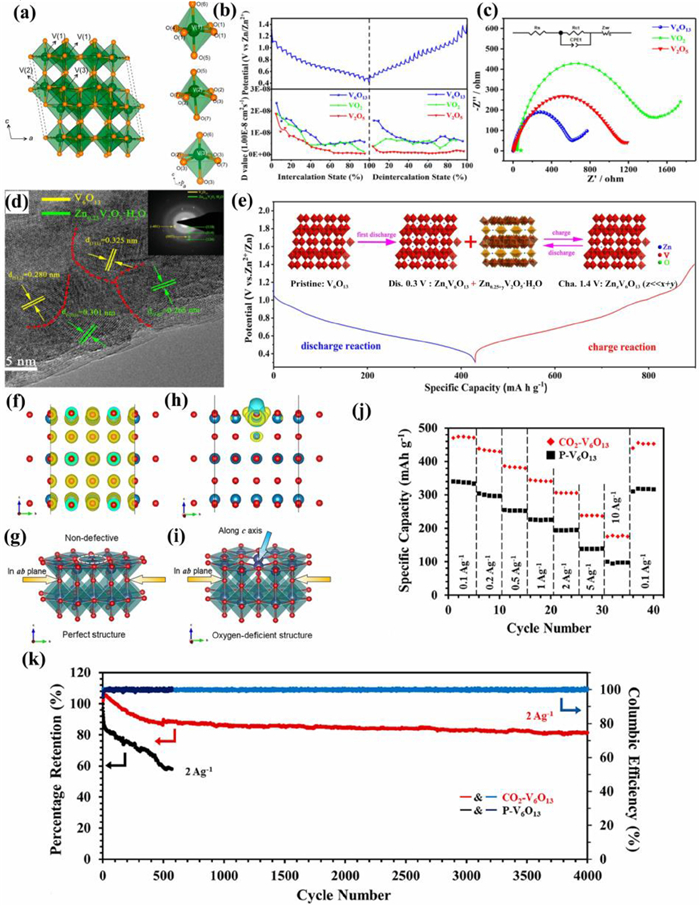
The trigonal phase V2O3 (R3c) has a 3D framework structure, which allows 3d electrons to move back and forth through the V-V chain followed by displaying metallic behavior. V atoms occupy two-thirds of octahedral interstices of O atoms, meanwhile two [VO6] octahedrons link by sharing corner, face and edge with the adjacent one to form a tunnel-like 3D structure (0.288 × 0.291 × 0.291 × 0.410 nm4 along the (111) lattice plane). These tunnel structures are conducive to the insertion/extraction of metal ions, so V2O3 materials are often used as electrode materials in electrochemical energy storage. Ding et al. [43] firstly applied porous V2O3 pyrolyzed from vanadium-based MOFs precursors as cathode of AZIBs. The cyclic voltammetry (CV) plot in Fig. 2a shows two coupled redox peaks at 0.92/1.04 and 0.56/0.72 V, which involved the two-step de/insertion reaction of Zn2+. This electrode displays a specific capacity of 300 mAh/g at 100 mA/g calculated based on Fig. 2b and also exhibits outstanding cycling stability with a capacity retention of 90% after 4000 cycles at 5 A/g (Fig. 2c). Ex-situ X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), and Raman tests were adopted to analyze the Zn2+-storage mechanism. In the discharge cycle, H2O molecules along with Zn2+ were migrated into the tunnel structures of V2O3 and then were leftover in the charge process, which would help to expand the ion diffusion channel and reduce the diffusion resistance of Zn2+ (Fig. 2d). The first-principle calculation further confirmed that the intercalated-Zn2+ occupied the octahedral interstices of O atoms to form a stable structure (Figs. 2e and f). The differential charge state density in Fig. 2g showed a chemical bond formation between Zn2+ and O2− from V2O3. The inserted Zn2+ reduced the distance and strengthened the interaction between the two atoms, resulting in the improved cycle stability of AZIBs. However, the octahedral interstice was not large enough for more than one zinc ion storage, which was confirmed by the corresponding calculation results. Subsequently, Ding et al. [44] found that the vanadium vacancies could also facilitate the Zn2+ intercalation/extraction tested via operando XRD and Raman analysis. Defect-rich V2O3 obtained from V2O5 nanosheets calcinated in NH3/Ar atmosphere by in-situ electrochemical lattice conversion reactions. Defect-rich V2O3 as cathode achieved a high capacity of 382.5 mAh/g and a remarkable rate performance. In the first charge process, the lattice structure of V2O3 was distorted to obtain vanadium vacancies, which were favorable for the Zn2+ de/intercalation in the subsequent cycles. The activated V2O3 by fabricating the vanadium vacancies could fasten the reaction kinetics of AZIBs, but the detailed reaction mechanism was still unclear. Luo et al. [45] investigated the reaction mechanism of fastening the electrochemical kinetics by preparing hollow-carved carbon-coated V2O3 microcuboids with abundant channels, short ion diffusion distance and good electrical conductivity (Fig. 2h). The electrolyte concentration played an important role in the electrochemical performance of V2O3 electrodes. The capacity and cycle stability in 2 mol/L Zn(CF3SO3)2 were superior to those of 1, 3 and 4 mol/L. The maximum capacity, high rate performance and capacity retention in 2 mol/L electrolyte was 625 mAh/g at 0.1 A/g, 486 mAh/g at 20 A/g, and 100% after 10,000 cycles, respectively (Fig. 2i). H2O as reductant provided the electron to V2O3 to form V10O24·12H2O (Fig. 2j) in the initial charge process, and then layer Zn3+x(OH)2+3xV2−xO7−3x·2H2O appeared in the subsequent discharge process. V10O24·12H2O and Zn3+x(OH)2+3xV2−xO7−3x·2H2O gradually were converted into V2O5·nH2O and ZnxV2O5·nH2O, respectively after 20th charging/discharging.

|
Download:
|
| Fig. 2. (a) CV at 0.2 mV/s between 0.3 V and 1.5 V and (b) charge/discharge curves (GCD) for the initial three cycles of the porous V2O3@C microsphere (P-V2O3@C). (c) Long-term cycling performance at 5 A/g. (d) Schematic diagram of Zn2+ storage mechanism, structure of V2O3 without (e) and with (f) Zn2+ being inserted. (g) Differential charge state density between V2O3 with the inserted Zn ions. Reproduced with permission [43]. Copyright 2019, American Chemical Society. (h) The preparation schematic illustration of the hierarchical carbon-coated V2O3. (i) Long-cycling performance of V2O3 in 2 mol/L Zn(CF3SO3)2. (j) The oxidation schematic illustration of V2O3 during the first charging. Reproduced with permission [45]. Copyright 2020, American Chemical Society. | |
VO2 phase with different space groups (B (C2/m), M (P2), A (P4), D (P2/c)) has been widely studied as the cathode material for AZIBs. The [VO6] octahedron as the basic structural unit of the above-mentioned four phases links by edge or vertice to form a tunnel structure. The two octahedrons of VO2(B) connect along the b-axis direction and the second layer moves (1/2, 1/2, 0) (fractional coordinates in unit cell) relative to the first layer, shaping into a tunnel structure. The V-V atoms in monoclinic VO2(M) form a zigzag-chain structure along the c axis, causing a staggered transverse displacement of vanadium atoms and an oxygen octahedron distortion. Hence, VO2(M) has denser tunnels than VO2(B). Similar to the VO2(M) structure, VO2(D) is built based on one chain composed of alternating the edge-sharing distorted [V(1)O6] octahedron linking to another individual chain composed of [V(2)O6] octahedron through corner-shared oxygen atoms. For VO2(A), four sets of two-sided octahedrons on the c plane form a 2 × 2 square. These squares stack along the c axis by edge-sharing to form long Z-shaped V-atom chains.
VO2(B) is suitable for use as the electrode materials of AZIBs due to higher theoretical capacity and the tunnel structure with plus sizes (0.34, 0.82 and 0.5 nm3 along a-, b- and c-axes, respectively) for the Zn2+ migration. Ding et al. [46] investigated the interaction behavior of Zn2+ with VO2(B) in AZIBs assembled with VO2(B) nanofibers as cathode, Zn foil as anode, and Zn(CF3SO3)2 as electrolyte (Fig. 3a). VO2(B) cathode displayed a specific capacity of 357 mAh/g (Fig. 3b) and superior rate capability (171 mAh/g at 51.2 A/g in Fig. 3c), ascribing to the intercalation pseudocapacitance behavior and the ultrafast kinetics of Zn2+ into the tunnel structures of VO2(B). The lattice parameter variation with the Zn2+ intercalation into the tunnels (paralleled to the b- and c-axes of VO2(B)) tested by in-situ XRD diffraction was less, suggesting that these tunnels were stable and large enough to ensure the fast Zn2+ intercalation/de-intercalation (Fig. 3d). Therefore, VO2(B) cathode with tunnel-structure was proved feasible to permit the insertion/extraction of Zn2+ in laboratory-scale research. Park et al. [47] further revealed the electrochemical reaction mechanism of AZIBs using a bond-valence sum energy map and a first-principle calculation. Among the four sites (Znc, ZnA1, ZnA2, Znc') of inserting Zn2+ into VO2(B) structure shown in Fig. 3e, ZnA2 site was the optimal location of Zn2+ into VO2(B) and the predicted redox potential of ZnxVO2(B) was ~0.61 V (Fig. 3f), close to the experimental value of ~0.7 V (VO2(B) + 0.57Zn2+ + 1.14e− ↔ Zn0.57VO2(B), Fig. 3g). The pseudocapacitance contribution of Zn2+ intercalating into VO2(B) structure, evaluated using potentiodynamic electrochemical impedance spectroscopy (PDEIS) technique was negligible. But the strong Coulombic ion-lattice interaction between Zn2+ and VO2(B) structure slowed down the kinetics and further limited the improvement of electrochemical properties. Oxygen-deficient VO2 nanostructure could address the above-mentioned problem through providing the jumping charge transport sites via enlarging the b tunnels for fast zinc-ion diffusion. VO2 with high-concentration oxygen vacancies prepared by Li et al. [48] displayed a discharge specific capacity of 375 mAh/g at 100 mA/g and long-term cyclic stability with a retained capacity of 175 mAh/g at 5 A/g over 2000 cycles in 3 mol/L Zn(CF3SO3)2. However, in aqueous electrolytes, Zn2+ often exists in the form of Zn(H2O)62+, indicating that larger desolvation energy and insertion energy need to be overcome in the process of Zn2+ insertion. And the strong electrostatic repulsion between Zn2+ and host structure also causes poor diffusion kinetics and mild deformation in the host structure. So Li et al. [49] announced a H+ (de)insertion mechanism with less structure distortion of VO2(B) than the Zn2+ insertion mechanism. The VO2(B) cathode exhibited excellent electrochemical performances of the first discharge capacity (353 mAh/g at 1.0 A/g), a 75.5% capacity retention after 945 cycles at 3.0 A/g in 1 mol/L ZnSO4 (Fig. 3h). The whole reaction steps were shown in Fig. 3i. The generated H+ in the deposition process of Zn4(OH)6SO4·5H2O could insert into VO2(B) to contribute most of capacity by the first-principle analysis (Fig. 3j) and keep long-term cycle stability. Zhang et al. [50] further verified the proton insertion mechanism by the bond valence method using VO2(M) as cathode and ZnSO4 as electrolyte. While storing Zn2+ in VO2(D) phase, H+ and Zn2+ could simultaneously insert into and extract from VO2(D) during the cycling process, resulting in a phase transition from VO2(D) to V2O5·xH2O [51]. H2O molecules with Zn2+ co-intercalation into host structure facilitated the intercalation of Zn2+ due to shielding the electrostatic interactions between charge carrier and VO2(D).

|
Download:
|
| Fig. 3. (a) Schematic of AZIB with VO2(B) nanowire as cathode and Zn foil as anode. (b) Cycling performance of VO2(B) cathode at 0.25 C (additional: GCD curve). (c) Rate capabilities of VO2(B) nanofibers. (d) Schematic view of Zn2+ insertion/extraction VO2(B) nanofibers projected along the direction of the b and c axes, respectively. Reproduced with permission [46]. Copyright 2018, Wiley-VCH. (e) Bond-valence sum energy map of VO2(B) for the a-b, b-c, and a-c planes showing all possible Zn-ion sites in the VO2(B) structure. (f) Formation energy of ZnxVO2(B) (0 ≤ x ≤ 0.5). (g) Calculated average redox potential and experimentally measured charge/discharge curve of ZnxVO2(B). Reproduced with permission [47]. Copyright 2018, American Chemical Society. (h) Cycling performance and the corresponding coulombic efficiency of VO2 nanorods under different current densities. (i) Diagram of electrochemical mechanism during discharge of Zn/VO2 battery. (j) The sequential insertion of H+ into the VO2. Reproduced with permission [49]. Copyright 2019, Wiley-VCH. | |
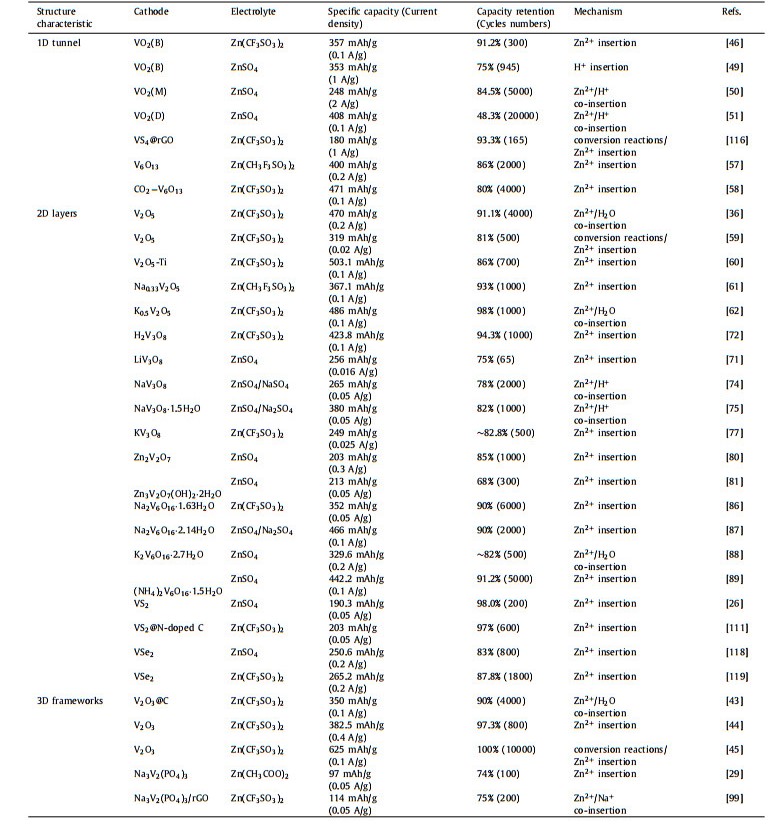
The monoclinic V6O13 (C2/m) with mixed-valence vanadium (V4+/V5+) consists of alternating single- and double-layer distorted [VO6] octahedrons connected by sharing corner to form chains zigzag along the b-axis [52], which can provide more active sites for ion storage (such as Li+ capacity: 417 mAh/g) [53, 54]. As shown in Fig. 4a, the valence states of V(1) atoms in single-layer and V(3) atoms in double-layer are +4, while the valence state of V(2) atoms in the double layer is +5. So the ratio of V4+/V5+ is 2:1 and an average mixed vanadium valence is V4.33+ [55]. Moreover, V6O13 at room temperature has a metallic character.

|
Download:
|
| Fig. 4. (a) Crystal structure of monoclinic V6O13: Three crystallographically nonequivalent vanadium sites in V6O13 are shown separately. Reproduced with permission [55]. Copyright 2017, American Chemical Society. (b) The charge/discharge galvanostatic intermittent titration technique (GITT) curves of V6O13 and comparison of Zn2+ diffusion coefficient. (c) Electrochemical impedance spectroscopy (EIS) results of V6O13, VO2, and V2O5. (d) The HRTEM image with the corresponding selected area electron diffraction (SAED) pattern at the fully discharged state. (e) Schematic diagram of the highly reversible phase transition during the discharge/charge process of V6O13. Reproduced with permission [56]. Copyright 2019, Wiley-VCH. (f, g) The charge distribution and corresponding structure of pristine V6O13. (h, i) The charge distribution and corresponding structure of oxygen-deficient V6O13. Reproduced with permission [57]. Copyright 2019, Wiley-VCH. (j) Rate performance for P-V6O13 (V6O13 with H2O only) and CO2-V6O13 (V6O13 with a mixture of H2O and CO2). (k) Cycling stability and coulombic efficiency for CO2-V6O13 and P-V6O13 at 2 A/g. Reproduced with permission [58]. Copyright 2021, American Chemical Society. | |
Shan et al. [56] prepared V6O13 cathode and investigated the Zn2+ storage mechanism in AZIBs. The V6O13 electrode displayed a specific capacity of 206 mAh/g at 10 A/g after 3000 cycles and an excellent rate performance owning to higher Zn2+ ion diffusion coefficient, better electronic conductivity than V2O5 and VO2 (Figs. 4b and c). Interestingly, the highly-reversible Zn0.25V2O5·H2O phase promoted the storage of Zn2+ verified by ex-situ XRD, high-resolution transmission electron microscope (HRTEM) and XPS (Fig. 4d). Fig. 4e illustrates the Zn2+ insertion/extraction mechanism. During the discharge process of aqueous Zn/V6O13 cell, Zn2+ is inserted into the empty sites of V6O13 phase to form a new Zn0.25V2O5·H2O phase; while in the subsequent charge process, Zn2+ is extracted from V6O13 and Zn0.25V2O5·H2O phase simultaneously disappears. A structural engineering strategy of oxygen-deficient is usually adopted to fasten the kinetics and boost the Zn2+ diffusion pathways for the reversibility and capacity of V6O13 cathode. As an example, the oxygen-deficient V6O13 structure was fabricated by a two-step method from solution-redox-based self-assembly method to thermal treatment under a reducing N2/H2 atmosphere, which was explored to create more Zn2+ intercalating sites [57]. The resulting V6O13 cathode retained a high specific capacity of 400 mAh/g after 200 cycles, reaching a utilization rate of 95% of the theoretical specific capacity. Its capacity retention still remained 86% after 2000 cycles because the vacancies of V6O13 provided more pathways of Zn2+. The surface electron density difference analysis proved that the charge distributions of oxygen-deficient V6O13 structure in Figs. 4h and i were more concentrated than those of perfect V6O13 in Figs. 4f and g, resulting in the dual-direction Zn2+ insertion formation and the capacity increase. As we all know, the strong electrostatic attraction between Zn2+ and host structure could hinder the Zn2+ diffusion, so CO2-trapped in V6O13 was designed to modify the electrochemical performance [58]. The as-prepared CO2-V6O13 electrode showed a maximum capacity of 471 mAh/g (Fig. 4j) and an excellent cyclic stability of 80% capacity retention after 4000 cycles at 2 A/g (Fig. 4k).
2.1.4. Vanadium pentoxide (V2O5)The layered orthorhombic V2O5 structure (Pmmn) is build-up of zigzag double chains of edge and corner sharing distorted [VO5] square pyramids. The layers are connected along the c-axis by weak electrostatic interactions, which are readily available for the intercalation of Zn2+. The interlayer spacing of 0.577 nm in V2O5 is much larger than the radius of Zn2+ (0.074 nm), so it can deliver a high theoretical capacity of 589 mAh/g.
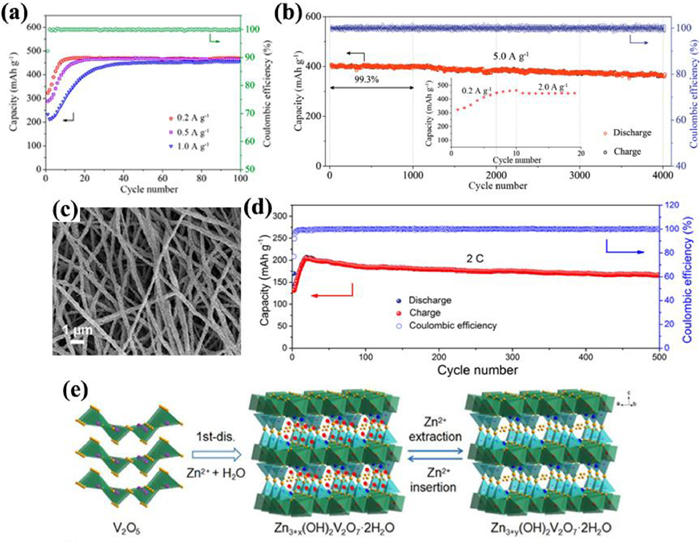
Zhang et al. [36] assembled AZIBs based on ball-milled commercial V2O5 cathode, Zn anode and 3 mol/L Zn(CF3SO3)2 electrolyte, whose reversible capacity reached 470 mAh/g at 0.2 A/g (Fig. 5a) and capacity retention was 91.1% over 4000 cycles at 5 A/g (Fig. 5b). Chen et al. [59] prepared porous V2O5 nanofibers (Fig. 5c) by electrospinning technology, which displayed the capacity of 319 mAh/g at 20 mA/g and capacity retention of 81% over 500 cycles at 2 C (Fig. 5d) in Zn(CF3SO3)2 electrolyte. A two-step conversion mechanism was adopted to explain the Zn2+ insertion/extraction (Fig. 5e): the open-structured zinc pyrovanadate (Zn3+x(OH)2V2O7·2H2O) formation in the first discharge and then the reversible insertion/extraction of Zn2+ between Zn3+x(OH)2V2O7·2H2O and Zn3+y(OH)2V2O7·2H2O. Optimizing electrode manufacture is an effective strategy to enhance the specific capacity of active materials. Javed et al. [60] directly grew 2D V2O5 nanosheets on Ti substrate to increase the contact between V2O5 and collector to avoid agglomeration, which as cathode exhibited a discharge capacity of 503.1 mAh/g at 100 mA/g and long-term stability with 86% retention after over 700 cycles at 500 mA/g.

|
Download:
|
| Fig. 5. (a) Galvanostatic cycling performance at 0.2, 0.5 and 1.0 A/g and the corresponding Coulombic efficiency at 0.2 A/g. (b) Long-cycling performance at 5.0 A/g (additional: the capacity evolution in the initial 19 cycles). Reproduced with permission [36]. Copyright 2018, American Chemical Society. (c) Scanning electron microscope (SEM) image of porous V2O5 nanofibers. (d) Long-term cycle performance of V2O5 cathode at 2 C. (e) Schematic of the reaction mechanism of V2O5 cathode. Reproduced with permission [59]. Copyright 2019, Elsevier. | |
The pre-intercalation of light alkali metal ions into V2O5 structure was also an effective strategy to improve the electrochemical properties through enlarging the interlayer distance. For instance, Na0.3V2O5 nanowire cathode had a high capacity of 367.1 mAh/g at 0.1 A/g and its capacity retention reached 93% after 1000 cycles in Zn//Na0.3V2O5 batteries [61]. The Na+ as "pillars" between V2O5 layers not only enhanced the conductivity of V2O5 but also improved the structural stability of V2O5 structure during the Zn2+ insertion/extraction. The incorporation of K+ into V2O5 layers adopted the Zn2+/H2O co-insertion storage mechanism. The K0.5V2O5 cathode showed excellent zinc storage performances with a reversible capacity of 397 mAh/g after 100 cycles at 1 A/g and 251 mAh/g after 1000 cycles at 5 A/g [62]. Even when the temperature was as low as –20 ℃, the capacity still reached to 241 and 115 mAh/g after 1000 cycles at 1 and 5 A/g, respectively. Density functional theory calculations further demonstrated that the introduction of K+ promoted the electron migration in the layer and thus boosted the (de)intercalation kinetics of Zn2+. In addition, there are other metal ions such as Li+ [63], Al3+ [64], Ag+ [65], Mg2+ [66], Zn2+ [67] and Ca2+ [68] to be investigated.
2.2. Introduction of vanadates in AZIBsVanadates as the derivatives of vanadium oxides are widely used in AZIBs owing to various cations. Cations in vanadates as pillars are inserted into the layers of different vanadium oxides to form a variety of vanadates (MxV3O8, MxV2O7, MxV6O16, and MxVyOz) with highly stable structure.
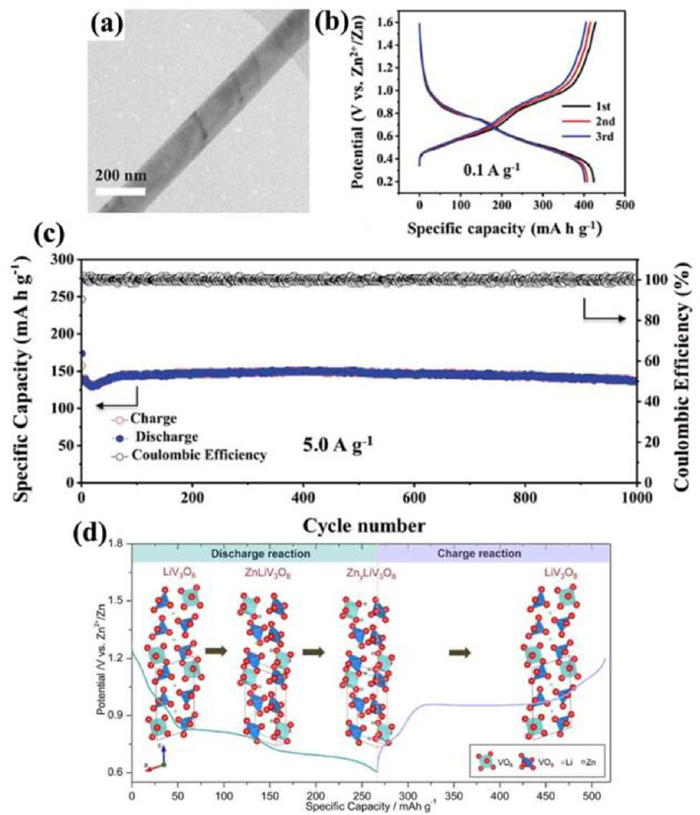
2.2.1. Main group trivanadates (MxV3O8)The monoclinic MxV3O8 structure is formed by the stacked [V3O8] layers and these layers are connected by M ions. Each [V3O8] layer consists of [VO6] octahedrons and [VO5] trigonal bipyramids. [VO6] octahedron links with [VO5] trigonal bipyramids by sharing vertice, resulting in an octahedral vacancy formation in the middle. M ions occupy the vacancy of [VO6] octahedron, in which ions are escaped uneasily [69-71]. Among them, H2V3O8 is an important member. H atoms between the layers link with O atoms in [VO6] polyhedron by a strong hydrogen bond, which can buffer the expansion/contraction of H2V3O8 structure during the ion insertion/extraction [70]. The ratio of V5+/V4+ is 2:1 in H2V3O8, which contributes to higher electrical conductivity than other vanadium oxides. For example, H2V3O8 nanowires (Fig. 6a) as cathode delivered a high initial capacity of 423.8 mAh/g at 0.1 A/g (Fig. 6b) and a capacity retention rate of 94.3% after 1000 cycles at 5 A/g (Fig. 6c) [72]. The large interlayer spacing of H2V3O8 contributed to an excellent Zn2+ storage performance. The similar phenomenon occurs in other monovalent ions (Li+, Na+, K+). LiV3O8, similar to the crystal structure of H2V3O8, also showed good electrochemical performance due to expanding the interlayer spacing [71]. ZnLiV3O8 transition phase formed at the early stage of discharge when Zn2+ were inserted into the Li-empty sites of LiV3O8 (Fig. 6d), and then ZnLiV3O8 transformed to ZnyLiV3O8 (y > 1) in the continuous intercalation of Zn2+. However, in the charging process, ZnyLiV3O8 was directly reduced to LiV3O8, suggesting that the intercalation/extraction of Zn2+ was reversible and Li+ was irreversible.

|
Download:
|
| Fig. 6. (a) Transmission electron microscopy (TEM) image of H2V3O8 nanowires. (b) Charge/discharge profiles at 0.1 A/g of the initial three cycles. (c) Long-term cycle performance of H2V3O8 cathode at 5 A/g. Reproduced with permission [72]. Copyright 2017, Wiley-VCH. (d) Schematic mechanism of Zn-intercalated LiV3O8 cathode. Reproduced with permission [71]. Copyright 2017, American Chemical Society. | |
The ion radius of Na+ (0.102 nm) is larger than those of H+ and Li+ (0.076 nm), so NaV3O8 with larger layer spacing (~0.71 nm) can promote ion transfer [73]. For instance, disordered NaV3O8 cathode exhibited a capacity of 265 mAh/g at 0.05 A/g in 3 mol/L ZnSO4 and 117 mAh/g at 0.4 A/g over 2000 cycles in 3 mol/L ZnSO4/0.5 mol/L Na2SO4 mixed electrolyte [74]. The storage behavior of Zn2+ was sequential two-stage intercalation of Zn2+ and H+ during discharging. If 1 mol/L Na2SO4 was added into 1 mol/L ZnSO4 solution, NaV3O8·1.5H2O nanoribbons could co-insert/extract of H+ and Zn2+, and then maintain a reversible capacity of 380 mAh/g at 0.05 A/g and a capacity retention of 82% over 1000 cycles at 4 A/g [75]. If 0.3 mol/L VOSO4 was added in 3 mol/L ZnSO4 electrolyte, fiber-like NaV3O8 cathode underwent a reversible three-ions insertion of VO2+, Zn2+ and H+ at the discharged state and displayed a high specific capacity of 450 mAh/g at 0.05 A/g and a capacity retention rate of 82% at 0.4 A/g after 500 cycles [76]. VO2+ carrier with large ion radius (0.16 nm) can still intercalate the layer of host structure and contribute the storage capacity. KV3O8 electrodes with an interlayer distance of ~0.77 nm delivered a discharge capacity of 182 mAh/g at 1.75 A/g with ~82.8% retention of initial capacity for 500 cycles at 1.25 A/g in 1 mol/L Zn(CF3SO3)2 electrolyte [77]. The pillar of K+ in host structure promoted an excellent capacity retention even at high rates due to its larger ion size.
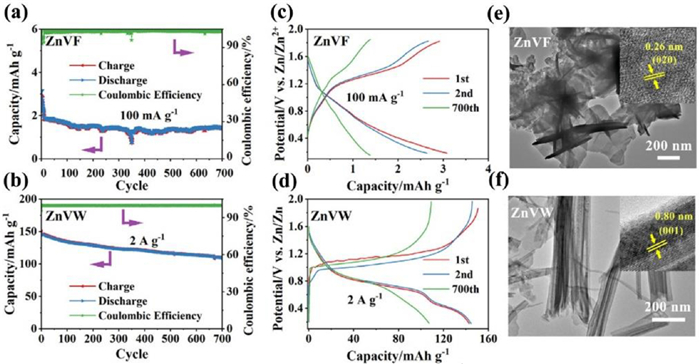
2.2.2. Main group polyvanadates (MxV2O7)The monoclinic α-Zn2V2O7 (C2/c) and trigonal Zn3V2O7(OH)2·2H2O (P3m1) phases are the typical representatives of MxV2O7. [V2O7] groups consist of a pair of [VO4] tetrahedrons both in α-Zn2V2O7 and Zn3V2O7(OH)2·2H2O structure, which align along c-axis to form the layer structure. [ZnO5] polyhedron in α-Zn2V2O7 is a distorted trigonal bipyramid and [ZnO6] polyhedron in Zn3V2O7(OH)2·2H2O is octahedron. The V-O-V pillars (tetrahedra) separate each Zn-O layer and H2O molecules can be randomly accommodated to ensure the facile diffusion of Zn ions [78-84]. The α-Zn2V2O7 nanowire displayed a high capacity retention of 85% after 1000 cycles at 4 A/g [80]. Its unique 1D nanowire morphology shortened the diffusion pathway of Zn2+ and electron transport distance because the growth direction of nanowires was parallel to the (002) plane (c-axis). Similarly, Zn3V2O7(OH)2·2H2O nanowires with porous framework prepared by a simple microwave method improved the intercalation of Zn2+ [81] and delivered the capacities of 213 and 76 mAh/g at 50 and 3 A/g, respectively, as well as good cycling stability. Cao et al. compared the electrochemical performances of 1D nanowires (ZnVW) and 2D nanoflakes (ZnVF) for Zn3V2O7(OH)2·2H2O (Figs. 7a–f) [78]. The nanowire cathode achieved more excellent discharge capacity of 108 mAh/g after 700 cycles at 2 A/g than that of the nanoflake cathode due to lower polarization, higher electrochemical activity, and faster ion diffusion capability of 1D materials. Hence, the electrochemical performances of MxV2O7 are highly dependent on preparation methods that influenced its morphology, particle size, and crystallinity beside inherent crystal structure.

|
Download:
|
| Fig. 7. (a, b) Charge/discharge cycling profile and coulombic efficiency of ZnVF and ZnVW cathode. (c) Galvanostatic charge/discharge curves of the 1st, 2nd and 700th cycles of ZnVF at the current density of 100 mA/g and (d) ZnVW at the current density of 2 A/g. TEM images of ZnVF (e) and ZnVW (f) after cycling (insets are HRTEM images). Reproduced with permission [78]. Copyright 2021, Elsevier. | |
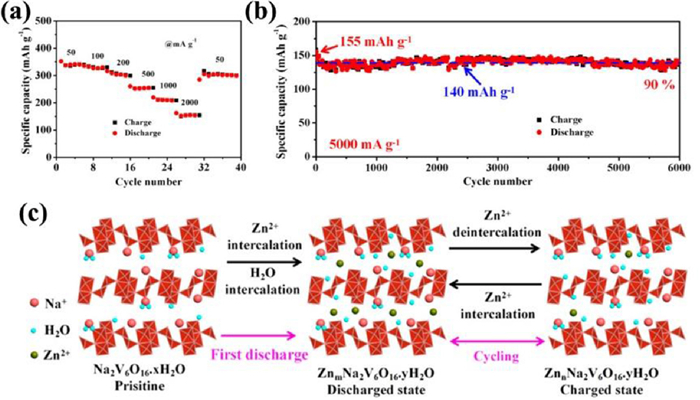
The monoclinic MxV6O16·nH2O comprises [V3O8] layers, which are constructed by edge-sharing [V2O8] square pyramids and [VO6] octahedrons, and interlayer hydrated M ions connect them together [85]. Much interest focuses on Na2V6O16·nH2O and K2V6O16·nH2O. Hu et al. [86] constructed 1D Na2V6O16·1.63H2O nanowire (Fig. 8a) by hydrothermal method with a high specific capacity of 352 mAh/g at 50 mA/g, a good rate performance and a high retention rate of 90% after 6000 cycles at 5 A/g (Fig. 8b). Zn2+ and H2O molecules were simultaneously inserted into the host structure in the first discharge (shown in Fig. 8c), and then only Zn2+ were reversibly inserted/extracted in the subsequent cycles. 1D Na2V6O16·1.63H2O nanobelt electrode adopted a similar Zn-storage mechanism and also exhibited superior electrochemical performance including a large discharge capacity of 466 mAh/g at 100 mA/g and excellent cycle stability with capacity retention of 90% over 2000 cycles at 20 A/g [87]. Sambandam et al. synthesized K2V6O16·2.7H2O nanorods, which as cathode achieved a reversible capacity of nearly 180 mAh/g at 6 A/g [88]. H2O molecules co-intercalated with Zn2+ as lubricant efficiently reduce the "effective charge" of Zn2+, thereby resulting in a higher diffusion coefficient of solvated Zn2+ and good electrochemical performance. 1D nanomaterials can offer more electrochemically active sites to promote the significant interfacial redox reaction. So the nanostructured morphologies of vanadium-based materials as desirable zinc ion hosts can contribute to improving their electrochemical properties.

|
Download:
|
| Fig. 8. (a) Rate performance of the Na2V6O16·1.63H2O at various current densities. (b) Cycling performances of the Na2V6O16·1.63H2O at 5000 mA/g. (c) Schematic of the reaction mechanism of Na2V6O16·1.63H2O cathode. Reproduced with permission [86]. Copyright 2018, American Chemical Society. | |
Compared to common intercalation metal ions, the existence of hydrogen bonds between NH4+ and [V3O8] layer offers a more stable layer structure, contributing to long-term cycling performance. Additionally, the lower molecular weight would provide higher gravimetric specific capacities theoretically. Zhang et al. [89] reported that there was the intensive hydrogen bond interaction between H atoms from H2O and NH4+ and O atoms from [V3O8] layer, which value was calculated to be 208.01 meV/Å2 by first-principles, higher than 49.58 meV/Å2 of V2O5, suppressing the dissolution of (NH4)2V6O16·1.5H2O. In the static immersion experiments of V2O5, V2O5·1.5H2O, NH4V3O8 and (NH4)2V6O16·1.5H2O cathodes, only (NH4)2V6O16·1.5H2O materials remained colorless after 200 days, suggesting the strong stability in the electrolyte. Furthermore, Wang et al. [90] presented (NH4)2V6O16·1.5H2O nanoribbons, which not only contributed to a high reversible capacity of 479 mAh/g at 0.1 A/g but also remained the specific capacity of 152 mAh/g at 5 A/g even after 3000 cycles. The results of ex-situ XRD, FTIR, XPS and HRTEM verified that the electrochemical reaction mechanism of (NH4)2V6O16·1.5H2O electrode was as follows: Zinc ions were intercalated into (NH4)2V6O16·1.5H2O along with a new phase of Zn3(OH)2V2O7·2H2O formation during the discharge process and were extracted from (NH4)2V6O16·1.5H2O accompanied by the new phase disappearing during the charging process.
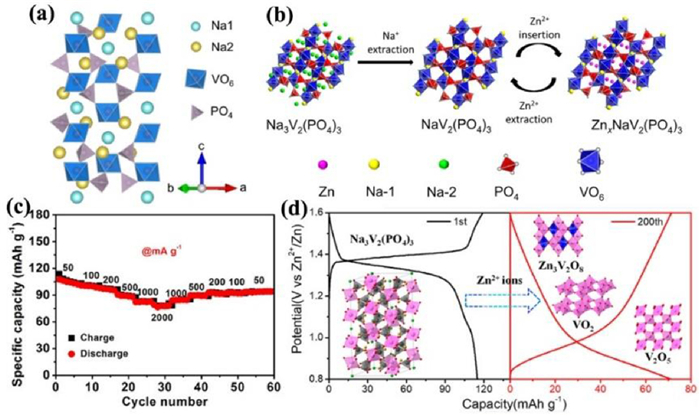
2.3. Introduction of alkali vanadium phosphates (MxV2(PO4)3, M = Li, Na) in AZIBsMxV2(PO4)3 (M = Li, Na) structure is composed of [VO6] octahedrons and [PO4] tetrahedrons by sharing all vertices to form the "lantern" [V2(PO4)3] unit framework, where M atoms occupy the voids of tetrahedrons and octahedrons (Fig. 9a). The open frame structure can endure the significant volume change (radius of Na+ (0.102 nm) and Li+ (0.076 nm) vs. Zn2+ (0.074 nm)) in the ion insertion or extraction process [91-97]. Monoclinic Li3V2(PO4)3 (P2, LVP) and rhombohedral Na3V2(PO4)3 (R3c, NVP) cathodes not only provide a robust structure but also a high working voltage (~1.8 V vs. Zn2+/Zn). Zhao et al. [98] compared the electrochemical mechanisms of Li3V2(PO4)3 (LVP) and Na3V2(PO4)3 (NVP) cathodes in mixed aqueous electrolyte (1 mol/L Li2SO4 and 2 mol/L ZnSO4). NVP cathode experienced Na+ and Li+ co-insertion while only two Li+ insertion for LVP cathode. Zn2+ precipitated on the surface of zinc metal, leading to initial discharge capacities of 128 and 96 mAh/g at 0.2 C for LVP and NVP cathodes, respectively. Compared with the low charge of Li+/Na+, Zn2+ with strong electrostatic interaction force was difficult to be inserted into the LVP/NVP lattice. However, Zn2+ could intercalate Na-deficit NVP structure (NaV2(PO4)3) through NVP to form ZnxNaV2(PO4)3 in the first discharge process and de-intercalate after charging shown in Fig. 9b [29], resulting in a reversible capacity of 97 mAh/g at 0.5 C and a capacity retention rate of 74% after 100 cycles in 0.5 mol/L Zn(CH3COO)2 electrolyte. If AZIBs assembled with NVP@reduced graphene oxide (NVP@rGO) microsphere cathode and 2 mol/L Zn(CF3SO3)2 electrolyte [99], Zn2+ and Na+ were simultaneously inserted into NVP structure tested by ex-situ XPS and XRD. Galvanostatic intermittent titration technique (GITT) test showed that the divalent Zn2+ had higher diffusion coefficient than monovalent Na+. The complete electrochemical reaction equation was as follows: NaxV2(PO4)3 + zZn2+ + yNa+ + (2z+y)e− ↔ ZnzNa(x+y)V2(PO4)3. The specific capacity of NVP@rGO cathode reached up to 74 mAh/g at 500 mA/g after 200 cycles and its rate performance was also excellent (107 mAh/g at 50 mA/g and 82 mAh/g at 2 A/g) (Fig. 9c). Ko et al. [100] proposed a quasi-two-stage Na+ and Zn2+ insertion mechanism based on the analysis results of synchrotron XRD and Rietveld refinement using porous NVP as cathode and 0.5 mol/L Zn(CH3COO)2 as electrolyte. The formation of Na-deficit NVP structure (NaV2(PO4)3) at the initial charging stage resulted in the predominant intercalation of Na+ followed by the Zn2+ intercalation into Na2 (18e site) place of NVP, which was more active than Na1(6b site) place. When discharged to 0.6 V, Zn0.25NaV2(PO4)3 phase with good electrochemical reversibility formed. However, the reason for Zn2+ occupying only Na2 site was still unknown. Hu et al. [101] revealed the Na+ and Zn2+ mixed occupation at both Na1 and Na2 sites of ZnxNaV2(PO4)3 based on experimental and theoretical methods, which improved the conductivity, crystal structure and structural stability of ZnxNaV2(PO4)3 after Zn2+ insertion. Na1 sites were activated due to the concerted migration of Na+/Zn2+. Recently, Li et al. [102] demonstrated that the NASICON-type compounds were not suitable for AZIB because of their structure decomposition during the long cycles (Fig. 9d) after comparing the structure stability of Na3V2(PO4)3 and Na3V2(PO4)2F3 cathodes in 1 mol/L Zn(CF3SO3)2. During cycling, Na3V2(PO4)3 decomposed into Zn3V2O8, V2O5, and V2O, while Na3V2(PO4)2F3 decomposed into V2O5, VPO5, and Zn3(OH)2V2O7·2H2O, greatly reducing the stability of NASICON-type compounds.

|
Download:
|
| Fig. 9. (a) Crystal structure of the NVP structure. Reproduced with permission [100]. Copyright 2020, American Chemical Society. (b) Schematic diagram for phase transition of Na3V2(PO4)3 cathode during cycling. Reproduced with permission [29]. Copyright 2016, Elsevier. (c) Rate performance of the NVP@rGO at various current densities. Reproduced with permission [99]. Copyright 2019, Elsevier. (d) The decomposition mechanism of Na3V2(PO4)3 during battery cycling. Reproduced with permission [102]. Copyright 2021, American Chemical Society. | |
Vanadium disulfide (VS2) is a representative member of transition metal dichalcogenide, which possesses the unique chain layered structure analogous to graphite. V atoms and S atoms in VS2 are covalently bonded, and neighboring layers (S-V-S) with a large interlayer distance of 0.58 nm are connected by weak van der Waals interaction, which can accommodate more the guest ion storage of Zn2+, Al3+ and Mg2+ [103-107]. Even if these guest ions are intercalated in the host structure, the structure of VS2 is not significantly broken [108, 109]. For instance, He et al. [26] firstly investigated the insertion mechanism of Zn2+ for VS2 nanosheet by means of in-situ Raman, ex-situ XRD and ex-situ XPS tests. The phase transitions from VS2 to Zn0.09VS2 and then to Zn0.23VS2 occurred within a voltage range of 0.65–0.82 and 0.45–0.65 V during the discharge process, respectively. The interlayer expansion of VS2 along (002) after Zn2+ inserting is only 1.73%. Hence VS2 nanosheet cathode for AZIBs provides a reversible specific capacity of 190.3 mAh/g at 0.05 A/g under the voltage window of 0.4–1.0 V. Even at 0.5 A/g, the first capacity retention rate reached 98% after 200 cycles. But in fact, the narrow voltage window (0.4–1.0 V vs. Zn2+/Zn) limited the energy density of VS2. Yu et al. [110] increased the working potential by in-situ fabricating heterostructural VS2/VOx materials. The average discharge voltage of VS2/VOx cathode was 0.9 V (vs. Zn/Zn2+), which is 0.25 V higher than that of the pristine VS2 cathode. So the composite electrode provided a reversible capacity of 150 mAh/g at 0.1 A/g. Even at 1 A/g, the capacity retention rate reached 75% after 3000 cycles. Liu et al. [111] adopted N-doped carbon as the conductive supports instead of VOx to improve the chemical stability and long-cycle performance with the help of strong interfacial interaction between VS2 and N-doped carbon. The resulting cathode displayed a high specific capacity of 203 mAh/g at 50 mA/g and long-term cycling stability with a capacity retention of 97% after 600 cycles at 1 A/g.
Monoclinic VS4 (I2/c) as an analog of VS2 is a unique 1D atomic-chain structure bonded by weak van der Waals forces, which is consisted of V4+ coordinated to sulfur dimers (S22−). VS4 with more S atoms shows larger interlayer space (0.583 nm) than that of VS2 [112-115]. Qin et al. [116] prepare VS4@rGO composite by the hydrothermal method, which displayed a capacity as high as 180 mAh/g at 1 A/g after 165 cycles and a capacity retention of 93.3%. During the battery charge/discharge, a conversion reaction occurred between VS4 and Zn3(OH)2V2O7·2H2O.
Hexagonal vanadium diselenide (VSe2, P3m1) is also a typical layered structure, where V atoms and Se atoms combine with covalent bonds to form the Se-V-Se sandwich structure. And the layers connect each other by van der Waals forces along c-axis direction. The electronegativity of Se is lower than those of O and S, which reduced the electrostatic interaction between the intercalation ions and the host structure and lowered the electron migration barrier [117]. Wang et al. [118] firstly presented VSe2 cathode with a large interlayer spacing of 0.61 nm and showed a discharge storage capacity of 250.6 mAh/g at 200 mA/g and a reversible specific capacity of 132.6 mAh/g even at a high current density of 5 A/g. Bai et al. [119] adopt defect engineering to fabricate the stainless steel (SS)-supported VSe2 nanosheets with defects to improve the conductivity and fasten the ion transmission speed (DZn2+ ≈ 10−8 cm2/s). Therefore, the synergistic effects of selenium defects and self-supporting structure improved the VSe2-x-SS structure stability and increased the specific capacity (265 mAh/g).
3. Conclusions and outlooksGenerally, the zinc-ion storage mechanisms of vanadium-based cathodes are the crystal structure-related theory. Table 1 lists the electrochemical performance and energy storage mechanism of vanadium-based cathodes. The storing Zn2+ ways of the monoclinic VO2, V6O13 and VS4 phases with 1D-tunnel structure heavily depend on different lattice types. The VO2(D) and VO2(M) cathodes with monoclinic-P lattice structure adopt the Zn2+/H+ co-insertion mechanism. The VO2(B) and V6O13 phases with monoclinic-C lattice structures are inclined to the Zn2+ insertion mechanism. However, in other monoclinic-C lattice structure (I2/c), VS4 cathode prefers to the conversion reactions and Zn2+ insertion mechanism.
|
|
Table 1 The typical vanadium-based cathodes and their electrochemical properties for AZIBs. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Most layered vanadium-based materials such as V2O5, VS2, VSe2, Zn3V2O7(OH)2·2H2O and Na2V6O16·2.14H2O provide 2D diffusion channels of facilitating Zn2+ insertion. The guest ions intercalated into layered V2O5-type compounds act as pillars to stabilize the layered structure, contributing more superior cycling performance than that of the tunnel structure in Table 1. Their expanding layer space can accommodate Zn2+ and H2O co-insertion. Moreover, H2O molecules not only act as a charge shielding medium to stabilize structure, but also reduce the "effective charge" of Zn2+ and promote the intercalation rate of Zn2+.
The framework-structured V2O3 and NASICON-type compounds have various interpenetrating 3D diffusion channels, which can accommodate more Zn2+. Hence the framework-structured cathodes with 3D tunnel can adopt all of the above-mentioned Zn-storage mechanisms. In addition, NASICON-type compounds can also permit Zn2+/Na+ co-insertion mechanism due to involving the Na+ de/intercalating.
The goal of this review is to summarize the recent progress of the vanadium-based compound family, including vanadium oxide, vanadates, alkali vanadium phosphates and other vanadium-based materials, and to present the relationship between the structural features and Zn2+ storage mechanisms, which are crucial for designing and preparing high-energetic materials. Although vanadium-based materials have made impressive progress in AZIBs cathodes, suitable anodes, electrolytes, current collectors and binders are equally important for the development of AZIBs. Hence, accelerating the development of high-performance AZIBs needs to pay enough attention to the following aspects.
(1) The dissolution of vanadium-based compounds in electrolytes during cycling causes the capacity fading and cycling stability reducing. Modifying electrolytes, constructing the integrated electrode and designing the cathodes with stable structure can address the above-mentioned shortcomings.
(2) The inherent low operating voltage of vanadium-based compounds hinders their practical applications. Introducing the electron-withdrawing groups on vanadium-based compounds and higher electronegative atom substitution or exploring more advanced materials could increase the voltage platform.
(3) The mechanism for energy storage plays a crucial role in guiding the design of the cathode materials. Many energy storage mechanisms have been put forward, but they do not reach an agreement so far. Therefore, more and more high-resolution in-situ characterization technologies need to be applied to provide further proof to elucidate the real storage mechanisms, which is of great significance to the application of vanadium-based materials in AZIBs cathodes.
(4) The inherently-poor electrical conductivity of vanadium-based materials causes their distinct volume changes during cycling. The highly-conductive additives such as carbon-based materials are an effective strategy to address this problem. Beside, metal element doping modification and defect engineering can improve electrochemical performance by boosting the chemical environment of vanadium-based compounds.
(5) Vanadium-based compounds with layered structures are gradually damaged due to the repeated insertion/extraction of Zn2+. The design and synthesis of core-shell and hollow structures will help vanadium-based compounds to maintain the structural stability.
Declaration of competing interestWe have no conflicts of interest to declare.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. U1910210, U1810204 and 22004122), Research Foundation for the Returned Overseas in Shanxi Provence (No. 2020-048) and the Central Guidance on Local Science and Technology Development Fund of Shanxi Province (No. YDZJSX2021A021).
| [1] |
G. Crabtree, Science 366 (2019) 422-424. DOI:10.1126/science.aax0704 |
| [2] |
Q. Li, Y. Liu, S. Guo, et al., Nano Today 16 (2017) 46-60. DOI:10.1016/j.nantod.2017.08.007 |
| [3] |
X. Jia, C. Liu, Z.G. Neale, et al., Chem. Rev. 120 (2020) 7795-7866. DOI:10.1021/acs.chemrev.9b00628 |
| [4] |
A.J. Samson, K. Hofstetter, S. Bag, et al., Energy Environ. Sci. 12 (2019) 2957-2975. DOI:10.1039/c9ee01548e |
| [5] |
X.B. Cheng, R. Zhang, C.Z. Zhao, et al., Chem. Rev. 117 (2017) 10403-10473. DOI:10.1021/acs.chemrev.7b00115 |
| [6] |
Y.M. Chiang, Science 330 (2010) 1485-1486. DOI:10.1126/science.1198591 |
| [7] |
J.W. Choi, D. Aurbach, Nat. Rev. Mater. 1 (2016) 16013. DOI:10.1038/natrevmats.2016.13 |
| [8] |
J.B. Goodenough, K.S. Park, J. Am. Chem. Soc. 135 (2013) 1167-1176. DOI:10.1021/ja3091438 |
| [9] |
E. Hu, X.Q. Yang, Nat. Mater. 17 (2018) 482-483. DOI:10.1038/s41563-018-0085-6 |
| [10] |
F. Wang, O. Borodin, T. Gao, et al., Nat. Mater. 17 (2018) 543-549. DOI:10.1038/s41563-018-0063-z |
| [11] |
L. Chen, Y. Ruan, G. Zhang, et al., Chem. Mater. 31 (2019) 699-706. DOI:10.1021/acs.chemmater.8b03409 |
| [12] |
D. Xie, F. Hu, X. Yu, et al., Chin. Chem. Lett. 31 (2020) 2268-2274. DOI:10.1016/j.cclet.2020.02.052 |
| [13] |
L. Qian, W. Yao, R. Yao, et al., Adv. Funct. Mater. 31 (2021) 2105736. DOI:10.1002/adfm.202105736 |
| [14] |
X. Han, H. Leng, Y. Qi, et al., Chem. Eng. J. 431 (2022) 133931. DOI:10.1016/j.cej.2021.133931 |
| [15] |
T. Shoji, M. Hishinuma, T. Yamamoto, J. Appl. Electrochem. 18 (1988) 521-526. DOI:10.1007/BF01022245 |
| [16] |
X. He, H. Zhang, X. Zhao, et al., Adv. Sci. 6 (2019) 1900151. DOI:10.1002/advs.201900151 |
| [17] |
N. Liu, X. Wu, Y. Zhang, et al., Adv. Sci. 7 (2020) 2000146. DOI:10.1002/advs.202000146 |
| [18] |
D. Chao, W. Zhou, C. Ye, et al., Angew. Chem. Int. Ed. 58 (2019) 7823-7828. DOI:10.1002/anie.201904174 |
| [19] |
W. Sun, F. Wang, S. Hou, et al., J. Am. Chem. Soc. 139 (2017) 9775-9778. DOI:10.1021/jacs.7b04471 |
| [20] |
C. Xu, B. Li, H. Du, et al., Angew. Chem. Int. Ed. 51 (2012) 933-935. DOI:10.1002/anie.201106307 |
| [21] |
R.D. Corpuz, L.M. Juan-Corpuz, M.T. Nguyen, et al., Int. J. Mol. Sci. 21 (2020) 3113. DOI:10.3390/ijms21093113 |
| [22] |
N. Zhang, F. Cheng, Y. Liu, et al., J. Am. Chem. Soc. 138 (2016) 12894-12901. DOI:10.1021/jacs.6b05958 |
| [23] |
J. Cao, D. Zhang, X. Zhang, et al., Appl. Surf. Sci. 534 (2020) 147630. DOI:10.1016/j.apsusc.2020.147630 |
| [24] |
X. Guo, J. Zhou, C. Bai, et al., Mater. Today Energy 16 (2020) 100396. DOI:10.1016/j.mtener.2020.100396 |
| [25] |
W. Jiang, X. Xu, Y. Liu, et al., J. Alloy. Compd. 827 (2020) 154273. DOI:10.1016/j.jallcom.2020.154273 |
| [26] |
P. He, M.Y. Yan, G.B. Zhang, et al., Adv. Energy Mater. 7 (2017) 1601920. DOI:10.1002/aenm.201601920 |
| [27] |
V. Soundharrajan, B. Sambandam, S. Kim, et al., Nano Lett. 18 (2018) 2402-2410. DOI:10.1021/acs.nanolett.7b05403 |
| [28] |
L. Xu, Y. Zhang, J. Zheng, et al., Mater. Today Energy 18 (2020) 100509. DOI:10.1016/j.mtener.2020.100509 |
| [29] |
G. Li, Z. Yang, Y. Jiang, et al., Nano Energy 25 (2016) 211-217. DOI:10.1016/j.nanoen.2016.04.051 |
| [30] |
D. Kundu, P. Oberholzer, C. Glaros, et al., Chem. Mater. 30 (2018) 3874-3881. DOI:10.1021/acs.chemmater.8b01317 |
| [31] |
K. Lu, B. Song, Y. Zhang, et al., J. Mater. Chem. A 5 (2017) 23628-23633. DOI:10.1039/C7TA07834J |
| [32] |
C.D. Wessells, S.V. Peddada, R.A. Huggins, et al., Nano Lett. 11 (2011) 5421-5425. DOI:10.1021/nl203193q |
| [33] |
Y.J. Zhang, Y. Wang, L. Lu, et al., J. Power Sources 484 (2021) 229263. DOI:10.1016/j.jpowsour.2020.229263 |
| [34] |
R. Chen, R. Luo, Y. Huang, et al., Adv. Sci. 3 (2016) 1600051. DOI:10.1002/advs.201600051 |
| [35] |
H. Tang, Z. Peng, L. Wu, et al., Electrochem. Energy Rev. 1 (2018) 169-199. DOI:10.1007/s41918-018-0007-y |
| [36] |
N. Zhang, Y. Dong, M. Jia, et al., ACS Energy Lett. 3 (2018) 1366-1372. DOI:10.1021/acsenergylett.8b00565 |
| [37] |
F. Wan, Z. Niu, Angew. Chem. Int. Ed. 58 (2019) 16358-16367. DOI:10.1002/anie.201903941 |
| [38] |
N. Liu, X. Wu, L. Fan, et al., Adv. Mater. 32 (2020) 1908420. DOI:10.1002/adma.201908420 |
| [39] |
N. Liu, B. Li, Z. He, et al., J. Energy Chem. 59 (2021) 134-159. DOI:10.1016/j.jechem.2020.10.044 |
| [40] |
P.Y. Zavalij, M.S. Whittingham, Acta Crystallogr. 55 (1999) 627-663. DOI:10.1107/S0108768199004000 |
| [41] |
X. Xu, F. Xiong, J. Meng, et al., Adv. Funct. Mater. 30 (2020) 1904398. DOI:10.1002/adfm.201904398 |
| [42] |
B.Y. Tang, L.T. Shan, S.Q. Liang, et al., Energy Environ. Sci. 12 (2019) 3288-3304. DOI:10.1039/c9ee02526j |
| [43] |
Y. Ding, Y. Peng, S. Chen, et al., ACS Appl. Mater. Interfaces 11 (2019) 44109-44117. DOI:10.1021/acsami.9b13729 |
| [44] |
J.W. Ding, H.Y. Zheng, H.G. Gao, et al., Adv. Energy Mater. 11 (2021) 2100973. DOI:10.1002/aenm.202100973 |
| [45] |
H. Luo, B. Wang, F. Wang, et al., ACS Nano 14 (2020) 7328-7337. DOI:10.1021/acsnano.0c02658 |
| [46] |
J. Ding, Z. Du, L. Gu, et al., Adv. Mater. 30 (2018) 1800762. DOI:10.1002/adma.201800762 |
| [47] |
J.S. Park, J.H. Jo, Y. Aniskevich, et al., Chem. Mater. 30 (2018) 6777-6787. DOI:10.1021/acs.chemmater.8b02679 |
| [48] |
Z. Li, Y. Ren, L. Mo, et al., ACS Nano 14 (2020) 5581-5589. DOI:10.1021/acsnano.9b09963 |
| [49] |
Z. Li, S. Ganapathy, Y. Xu, et al., Adv. Energy Mater. 9 (2019) 1900237. DOI:10.1002/aenm.201900237 |
| [50] |
L. Zhang, L. Miao, B. Zhang, et al., J. Mater. Chem. A 8 (2020) 1731-1740. DOI:10.1039/c9ta11031c |
| [51] |
L. Chen, Z. Yang, Y. Huang, Nanoscale 11 (2019) 13032-13039. DOI:10.1039/c9nr03129d |
| [52] |
P. Kiri, G. Hyett, R. Binions, Adv. Mater. Lett. 1 (2010) 86-105. DOI:10.5185/amlett.2010.8147 |
| [53] |
N.A. Chernova, M. Roppolo, A.C. Dillon, et al., J. Mater. Chem. 19 (2009) 2526-2552. DOI:10.1039/b819629j |
| [54] |
Y.L. Ding, Y. Wen, C. Wu, et al., Nano Lett. 15 (2015) 1388-1394. DOI:10.1021/nl504705z |
| [55] |
W. Meng, R. Pigliapochi, P.M. Bayley, et al., Chem. Mater. 29 (2017) 5513-5524. DOI:10.1021/acs.chemmater.7b00428 |
| [56] |
L. Shan, J. Zhou, W. Zhang, et al., Energy Technol. 7 (2019) 1900022. DOI:10.1002/ente.201900022 |
| [57] |
M. Liao, J. Wang, L. Ye, et al., Angew. Chem. Int. Ed. 59 (2020) 2273-2278. DOI:10.1002/anie.201912203 |
| [58] |
W. Shi, B. Yin, Y. Yang, et al., ACS Nano 15 (2021) 1273-1281. DOI:10.1021/acsnano.0c08432 |
| [59] |
X. Chen, L. Wang, H. Li, et al., J. Energy Chem. 38 (2019) 20-25. DOI:10.1016/j.jechem.2018.12.023 |
| [60] |
M.S. Javed, H. Lei, Z. Wang, et al., Nano Energy 70 (2020) 104573. DOI:10.1016/j.nanoen.2020.104573 |
| [61] |
P. He, G.B. Zhang, X.B. Liao, et al., Adv. Energy Mater. 8 (2018) 1702463. DOI:10.1002/aenm.201702463 |
| [62] |
G. Su, S. Chen, H. Dong, et al., Nanoscale 13 (2021) 2399-2407. DOI:10.1039/d0nr07358j |
| [63] |
L.L. Fan, Z.H. Li, W.M. Kang, ACS Sustain. Chem. Eng. 9 (2021) 5095-5104. DOI:10.1021/acssuschemeng.0c09264 |
| [64] |
Q. Pang, W. He, X.Y. Yu, et al., Appl. Surf. Sci. 538 (2021) 148043. DOI:10.1016/j.apsusc.2020.148043 |
| [65] |
L.T. Shan, Y.Q. Yang, W.Y. Zhang, et al., Energy Storage Mater. 18 (2019) 10-14. DOI:10.1016/j.ensm.2018.08.008 |
| [66] |
F. Ming, H. Liang, Y. Lei, et al., ACS Energy Lett. 3 (2018) 2602-2609. DOI:10.1021/acsenergylett.8b01423 |
| [67] |
L. Wang, K.W. Huang, J. Chen, et al., Sci. Adv. 5 (2019) eaax4279. DOI:10.1126/sciadv.aax4279 |
| [68] |
C. Xia, J. Guo, P. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 3943-3948. DOI:10.1002/anie.201713291 |
| [69] |
H. Li, T. Zhai, P. He, et al., J. Mater. Chem. 21 (2011) 1780-1787. DOI:10.1039/C0JM02788J |
| [70] |
Y. Oka, T. Yao, N. Yamamoto, J. Solid State Chem. 89 (1990) 372-377. DOI:10.1016/0022-4596(90)90279-7 |
| [71] |
M.H. Alfaruqi, V. Mathew, J. Song, et al., Chem. Mater. 29 (2017) 1684-1694. DOI:10.1021/acs.chemmater.6b05092 |
| [72] |
P. He, Y. Quan, X. Xu, et al., Small 13 (2017) 1702551. DOI:10.1002/smll.201702551 |
| [73] |
Y.S. Cai, F. Liu, Z.G. Luo, et al., Energy Storage Mater. 13 (2018) 168-174. DOI:10.1016/j.ensm.2018.01.009 |
| [74] |
X. Shan, S. Kim, A.M.M. Abeykoon, et al., ACS Appl. Mater. Interfaces 12 (2020) 54627-54636. DOI:10.1021/acsami.0c15621 |
| [75] |
F. Wan, L. Zhang, X. Dai, et al., Nat. Commun. 9 (2018) 1656. DOI:10.1038/s41467-018-04060-8 |
| [76] |
S. Kim, X. Shan, M. Abeykoon, et al., ACS Appl. Mater. Interfaces 13 (2021) 25993-26000. DOI:10.1021/acsami.1c04279 |
| [77] |
H.J. Kim, J.H. Jo, J.U. Choi, et al., J. Power Sources 478 (2020) 229072. DOI:10.1016/j.jpowsour.2020.229072 |
| [78] |
H. Cao, C. Peng, Z. Zheng, et al., Electrochim. Acta 388 (2021) 138646. DOI:10.1016/j.electacta.2021.138646 |
| [79] |
Z. Pan, J. Yang, J. Yang, et al., ACS Nano 14 (2020) 842-853. DOI:10.1021/acsnano.9b07956 |
| [80] |
B. Sambandam, V. Soundharrajan, S. Kim, et al., J. Mater. Chem. A 6 (2018) 3850-3856. DOI:10.1039/C7TA11237H |
| [81] |
C. Xia, J. Guo, Y. Lei, et al., Adv. Mater. 30 (2018) 1705580. DOI:10.1002/adma.201705580 |
| [82] |
D. Diaz-Anichtchenko, D. Santamaria-Perez, T. Marqueno, et al., J. Alloy. Compd. 837 (2020) 155505. DOI:10.1016/j.jallcom.2020.155505 |
| [83] |
J. Guo, J. Ming, Y. Lei, et al., ACS Energy Lett. 4 (2019) 2776-2781. DOI:10.1021/acsenergylett.9b02029 |
| [84] |
A. Bayat, A.R. Mahjoub, M.M. Amini, J. Mater. Sci. Mater. Electron. 29 (2017) 2915-2926. |
| [85] |
K. Zhu, T. Wu, K. Huang, Adv. Energy Mater. 9 (2019) 1901968. DOI:10.1002/aenm.201901968 |
| [86] |
P. Hu, T. Zhu, X. Wang, et al., Nano Lett. 18 (2018) 1758-1763. DOI:10.1021/acs.nanolett.7b04889 |
| [87] |
F. Hu, D. Xie, D. Zhao, et al., J. Energy Chem. 38 (2019) 185-191. DOI:10.1016/j.jechem.2019.03.036 |
| [88] |
B. Sambandam, V. Soundharrajan, S. Kim, et al., J. Mater. Chem. A 6 (2018) 15530-15539. DOI:10.1039/C8TA02018C |
| [89] |
L. Zhang, J. Hu, B. Zhang, et al., J. Mater. Chem. A 9 (2021) 7631-7639. DOI:10.1039/d1ta00263e |
| [90] |
X. Wang, B. Xi, Z. Feng, et al., J. Mater. Chem. A 7 (2019) 19130-19139. DOI:10.1039/c9ta05922a |
| [91] |
E. Boivin, J.N. Chotard, C. Masquelier, et al., Molecules 26 (2021) 1428. DOI:10.3390/molecules26051428 |
| [92] |
I.V. Zatovsky, Acta Crystallogr. E66 (2010) i12. DOI:10.1107/S1600536810002801 |
| [93] |
S. Liu, L. Kang, J.M. Kim, et al., Adv. Energy Mater. 10 (2020) 2000477. DOI:10.1002/aenm.202000477 |
| [94] |
Q. Wang, J. Xu, W. Zhang, et al., J. Mater. Chem. A 6 (2018) 8815-8838. DOI:10.1039/C8TA01627E |
| [95] |
S.Y. Lim, H. Kim, R.A. Shakoor, et al., J. Electrochem. Soc. 159 (2012) A1393-A1397. DOI:10.1149/2.015209jes |
| [96] |
J. Zhang, Y. Fang, L. Xiao, et al., ACS Appl. Mater. Interfaces 9 (2017) 7177-7184. DOI:10.1021/acsami.6b16000 |
| [97] |
S. Boudin, A. Guesdon, A. Leclaire, et al., Int. J. Inorg. Mater. 2 (2000) 561-579. DOI:10.1016/S1466-6049(00)00074-X |
| [98] |
H.B. Zhao, C.J. Hu, H.W. Cheng, et al., Sci. Rep. 6 (2016) 25809. DOI:10.1038/srep25809 |
| [99] |
P. Hu, T. Zhu, X. Wang, et al., Nano Energy 58 (2019) 492-498. DOI:10.1016/j.nanoen.2019.01.068 |
| [100] |
J.S. Ko, P.P. Paul, G. Wan, et al., Chem. Mater. 32 (2020) 3028-3035. DOI:10.1021/acs.chemmater.0c00004 |
| [101] |
P. Hu, Z. Zou, X. Sun, et al., Adv. Mater. 32 (2020) e1907526. DOI:10.1002/adma.201907526 |
| [102] |
W. Li, X. Jing, K. Jiang, et al., ACS Appl. Energy Mater. 4 (2021) 2797-2807. DOI:10.1021/acsaem.1c00067 |
| [103] |
C. Tan, H. Zhang, Chem. Soc. Rev. 44 (2015) 2713-2731. DOI:10.1039/C4CS00182F |
| [104] |
L. Wu, R. Sun, F. Xiong, et al., Phys. Chem. Chem. Phys. 20 (2018) 22563-22568. DOI:10.1039/c8cp04772c |
| [105] |
L. Li, Z. Li, A. Yoshimura, et al., Nat. Commun. 10 (2019) 1764. DOI:10.1038/s41467-019-09400-w |
| [106] |
X. Huang, Z. Zeng, H. Zhang, Chem. Soc. Rev. 42 (2013) 1934-1946. DOI:10.1039/c2cs35387c |
| [107] |
D. Yu, Q. Pang, Y. Gao, et al., Energy Storage Mater. 11 (2018) 1-7. DOI:10.1016/j.ensm.2017.09.002 |
| [108] |
J. Zhou, L. Wang, M. Yang, et al., Adv. Mater. 29 (2017) 1702061. DOI:10.1002/adma.201702061 |
| [109] |
J. Feng, X. Sun, C. Wu, et al., J. Am. Chem. Soc. 133 (2011) 17832-17838. DOI:10.1021/ja207176c |
| [110] |
D. Yu, Z. Wei, X. Zhang, et al., Adv. Funct. Mater. 31 (2020) 2008743. |
| [111] |
J. Liu, W. Peng, Y. Li, et al., J. Mater. Chem. C 9 (2021) 6308-6315. DOI:10.1039/d1tc00531f |
| [112] |
S. Britto, M. Leskes, X. Hua, et al., J. Am. Chem. Soc. 137 (2015) 8499-8508. DOI:10.1021/jacs.5b03395 |
| [113] |
Y. Wang, Z. Liu, C. Wang, et al., Adv. Mater. 30 (2018) 1802563. DOI:10.1002/adma.201802563 |
| [114] |
Y. Zhou, Y. Li, J. Yang, et al., ACS Appl. Mater. Interfaces 8 (2016) 18797-18805. DOI:10.1021/acsami.6b04444 |
| [115] |
R. Sun, Q. Wei, Q. Li, et al., ACS Appl. Mater. Interfaces 7 (2015) 20902-20908. DOI:10.1021/acsami.5b06385 |
| [116] |
H. Qin, Z. Yang, L. Chen, et al., J. Mater. Chem. A 6 (2018) 23757-23765. DOI:10.1039/c8ta08133f |
| [117] |
M. Mao, X. Ji, S. Hou, et al., Chem. Mater. 31 (2019) 3183-3191. DOI:10.1021/acs.chemmater.8b05218 |
| [118] |
L.L. Wang, Z.X. Wu, M.J.H. Jiang, et al., J. Mater. Chem. A 8 (2020) 9313-9321. DOI:10.1039/d0ta01297a |
| [119] |
Y. Bai, H. Zhang, B. Xiang, et al., ACS Appl. Mater. Interfaces 13 (2021) 23230-23238. DOI:10.1021/acsami.1c04596 |