 2023, Vol. 34
2023, Vol. 34 


b Coordination Chemistry Institute, State Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, China
Over the past few decades, the progressively increasing emission of carbon dioxide (CO2) is speculated as the cause of global warming, sea-level rise along with subsequent other serious climate changes [1-3]. As a result, quite a lot of endeavors have been devoted to alleviating CO2 pollution, which would capture CO2 for conversion into reliable C1 feedstock of specific chemical reactions and acquire numerous desirable value-added chemicals [4, 5]. Amongst these reactions that incorporate CO2, the coupling reaction of CO2 with epoxides for the synthesis of cyclic carbonates is considered as one of the most popular strategies for reducing CO2 on account of widespread application of cyclic carbonates (chemical intermediates, electrolytes and aprotic polar solvents, etc.) and high atom utilization [6-8]. Nevertheless, industrial catalysts are generally homogeneous and leave certain limitations of product separation and catalyst recovery in the cycloaddition of CO2 and epoxides [9, 10]. Therefore, highly active and widely applicable heterogeneous catalysts need to be legitimately developed to apply in the CO2 fixation into cyclic carbonates. Very recently, polyoxometalates (POMs), as a representative of multifunctional crystalline materials, have been explored as catalyst in some reactions, such as cyclization reaction of 2-acylbenzoic acids with alcohols and the synthesis of sulfonyl pyrazoles [11-13].
Polyoxovanadates (POVs), as one great subclass of polyoxometalates, have been subject to extensive studies due to their aesthetically fascinating geometrical features, as well as their potential applications in magnetism, catalysis, and bioactivities, among others [14-20]. The emerging POVs, as long-promised heterogeneous candidate oxidative catalysts, have received continuous interest because of potential Lewis-acid active sites [21-26]. In particular, some representative works have been conducted by research groups of Hu, Wang, Wei, and so on, and the most renowned finding is that hybrid transition metal-POVs-based materials have been certified as effective heterogeneous catalysts for several reactions with good selectivity and effectiveness [21, 24-27]. For example, Huang and Yang et al. reported an imidazole-modified Ag-POVs framework, which can serve as efficient heterogeneous catalyst for detoxification of simulant sulfur mustard and construction of C—N bond [28]. But for vanadium oxide clusters, only quit a few reports involved in the catalytic application in the field of the oxidation sulfides and alcohols, and hardly involved in cycloaddition reactions [29-32]. Illuminated by the above viewpoints and our efforts in heterogeneous catalysis [30, 33, 34], there is still much room for exploring the catalytic properties of vanadium oxide clusters in the oxidation sulfides and cycloaddition reactions.
Herein, employing similar solvothermal synthetic approach, we elaborately designed and isolated four new hexanuclear polyoxovanadium clusters modified by 2-aminophenol and four different Lewis bases (LB), including ethanediamine (en), 1,2-diaminopropane, 1,2-cyclohexanediamine and N-(2-hydroxyethyl)ethylenediamine (ben) together: [(C6H6ON)2(C2H8N2)2(CH3O)6V6IVO8] (V6–1), [(C6H6ON)2(C3H10N2)2(CH3O)6V6IVO8] (V6-2), [(C6H6ON)2(C6H14N2)2(CH3O)6V6IVO8] (V6-3) and [(C6H6ON)2(C4H11N2O)2(CH3O)4V6IVO8] (V6-4). To our knowledge, the principal building unit, {V6O8} cluster, represents a brand-new configuration, which differs from other previously documented hexavanadate clusters. As expected, the open V sites in V6-1-4 make them be utilized as efficient Lewis acid catalysts with good stability for the cycloaddition reaction. Remarkably, the catalytic performance of V6–1 for heterogeneous oxidative desulfurization reaction also have been examined in this paper.
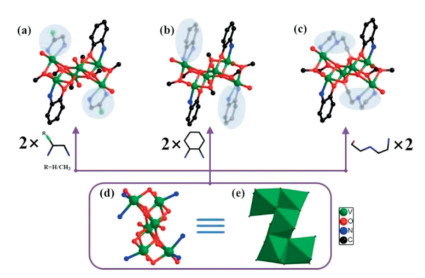
Single crystal X-ray diffraction (SCXRD) indicates that V6–1, V6-2 and V6-3 are isomorphic and crystallize in the monoclinic P21/n, monoclinic C2/c and orthorhombic Pbca space group, respectively (Figs. 1a and b, Table S1 in Supporting information). Thus, V6–1 was chosen as the representative to describe the structure. The basic structural unit of V6–1 is comprised of one independent [V6IVO8]8+, two [C6H6ON]−, two en molecules and six methoxy groups (Fig. 1a). The principal building unit is a distinct Z-shaped {V6O8} cluster (6.02 Å × 8.06 Å) (Fig. S2 in Supporting information), which can be described as the combination of one {V3O4} and another one that rotated 180° splice by edge-sharing mode (Figs. 1d and e). As shown in Fig. 1a, two en, two 2-aminophenol and six methoxy are decorated on the outer of the {V6O8} cluster. On the side, the principal building unit [V6IVO8]8+in V6-4 is same as that of V6-1-3 except that the number of methoxy groups reduces to four due to the tridentate ben ligand can link more than bidentate en molecules (Fig. 1c). Here, each V cation is hesacoordinated with a distorted octahedral geometry. And the oxidation states of all V centers were assigned as the VIV centers by bond valence sum (BVS) calculations (Tables S10-S13 in Supporting information).

|
Download:
|
| Fig. 1. (a) The ball-and-stick model of V6–1 and V6–2; (b) V6–3; (c) V6–4. (d, e) The ball-and-stick model and polyhedron of {V6} cluster. All H atoms were omitted for clarity. | |
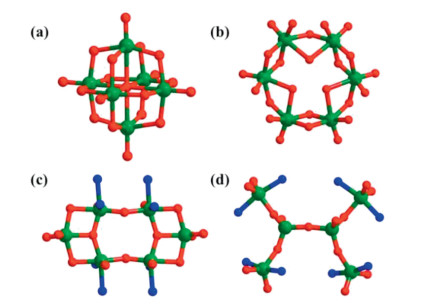
Remarkably, hexavanadate cluster, as the principal building unit, exhibits an unprecedented Z-shaped structure that differs from other previously reported oxovanadium clusters such as Lindqvist (Fig. 2a) [35-38], Anderson (Fig. 2b) [39], cyclic, and other structure topologies hexavanadate clusters (Figs. 2c and d) [32, 40, 41]. Hence, {V6O8} represents an important new member of hexanuclear vanadium clusters.

|
Download:
|
| Fig. 2. (a) Lindqvist-type, (b) Anderson-type, (c) cyclic and (d) other structure topologies hexanuclear vanadium clusters. | |
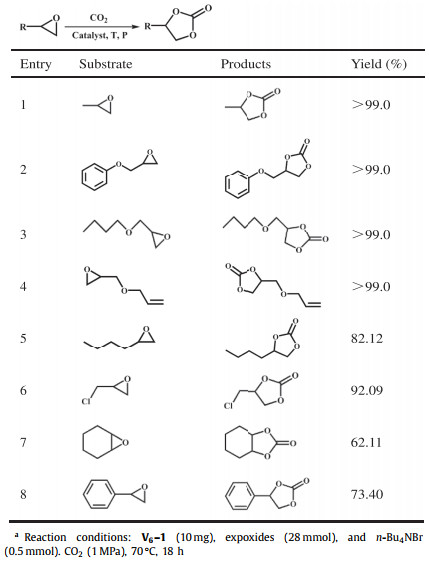
Considering that Z-shaped {V6} cluster with the exposed V sites could act as Lewis acid catalyst, V6–1 was employed in the CO2 fixation into cyclic carbonates reaction. To determine the optimum reaction conditions, propylene oxide (PO) was chosen as the substrate and V6–1 was used as a catalyst for preliminary investigations owing to the most abundance in terms of yield. Concretely, when 10 mg of V6–1 and 0.5 mmol of n-Bu4NBr participate in the reaction together, the conversion rate of PO gradually increases from 9.29% to > 99% with the growth of reaction pressure, temperature and time. After a series of contrast experiments were performed by altering reaction conditions, the optimal conditions were found to be entry 8 in Table S2 (Supporting information), that is, 10 mg catalyst loading with 0.5 mmol n-Bu4NBr as co-catalyst in 1 MPa pressure and for 18 h at 70 ℃, which allowed the conversion rate finally reaches > 99%. Similarly, V6-derivatives V6-2-4 also exhibited significantly enhanced catalytic properties (Fig. S19 in Supporting information). The final yield could not reach an expected value when reaction temperature, pressure and time are much lower. As illustrated in Table S2, a quite low yield was achieved when separately using only n-Bu4NBr (21.63%) or only V6–1 (11.51%) as catalyst (Table S2, entries 9 and 10), indicating that V6–1 and n-Bu4NBr play important role of synergistic catalysis during the reaction. Similarly, the yield of PO to the corresponding product was increased to 33.52% when catalyst V6–1 was replaced by equimolar raw materials NH4VO3 (Table S2, entry 12). In light of the above results, it can be generalized that V6–1 possessed distinct catalytic activity for the cycloaddition with epoxides for the synthesis of cyclic carbonates. In addition, the comparison of catalytic performance of V6–1 with several catalysts reported in the literature was also made (Table S3 in Supporting information).
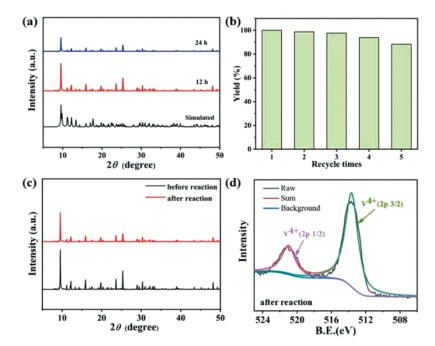
To further investigate the stability of V6–1 in the reaction system, we measured PXRD patterns of V6–1 immersed in PO at different durations (Fig. 3a). The characteristic peaks remained similar to that of simulated PXRD patterns, demonstrating that crystal samples possess pretty good stability in the reaction solution. Besides, V6–1 can be easily recovered from the reaction solution by filtering and washing with MeOH and then dried in air, and the conversion of the third repetition (98.4%) was reduced by only about 1.6% (Fig. 3b), which reveals that V6–1 still retains good catalytic performance after three cycles. It is worth noting that the following slight decline of yield could be related to mass loss of the catalyst. Subsequently, the consistent PXRD patterns (Fig. 3c), IR spectra (Fig. S20 in Supporting information) and X-ray photoelectron spectroscopy (XPS) spectra (Fig. 3d) of V6–1 after five cycles validate that its structural integrity after catalysis remained unchanged.

|
Download:
|
| Fig. 3. (a) PXRD patterns of V6–1 immersed in PO at different durations. (b) Recycling tests for the cycloaddition reaction using V6–1 as catalyst. (c) PXRD patterns of V6–1 before and after circulations. (d) XPS spectra of V 2p for V6–1 after circulations. | |
Serval expoxides as substrate were then selected for the cycloaddition reaction under the optimal conditions to assess the application scope of V6–1 as a catalyst. According to Table 1, 2-butyloxirane and epichlorohydrin achieved conversions of 82.12% and 92.09% after 18 h, respectively (Table 1, entries 5 and 6). Owing to the fact that the electron-withdrawing groups exist in the structure of substrates [42], the substrates glycidyl phenyl ether, 2-(butoxymethyl)oxirane and allyl glycidyl ether can be entirely converted to the corresponding products (Table 1, entries 2–4). However, for epoxycyclohexane and styrene oxide, only 62.11% and 73.40% substrates were transformed into the corresponding carbonates (Table 1, entries 7 and 8), which could be attributed to huge steric hindrance [43, 44]. Accordingly, we can draw a conclusion that the steric hindrance of substrates has a major influence on the conversion of expoxide to cyclic carbonate.
|
|
Table 1 Cycloaddition of CO2 with different substrates.a |
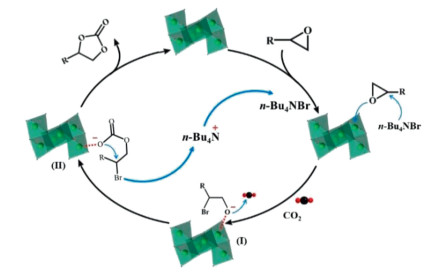
Based on the above results and previous work [45-47], we speculate a plausible catalytic mechanism for explaining the cycloaddition of CO2 to epoxide process, as shown in Scheme 1. First of all, the Lewis acidic V sites in the Z-shaped {V6O8} cluster are inclined to interact with the epoxide. Then the epoxy ring was activated and the related C–O bonds were weakened, which facilitated nucleophilic attack of the Br− on the carbon of the epoxy ring and further generated the ring-opening intermediate I. Meanwhile, the O anion of the intermediates I coupled with the CO2 molecules to produce a metal-carbonate intermediate II. Finally, the O atom of II attacks its carbonate anion to release the bromo, finishing the ring closure and giving the cyclic carbonate. According to the above results, it could be concluded that the "fusion" of n-Bu4NBr and V sites in V6–1 might synergistically impact the catalytic process, which played a considerable role in facilitating the resultant catalytic performance.

|
Download:
|
| Scheme 1. Plausible mechanism for the CO2 cycloaddition reaction over n-Bu4NBr/V6–1 catalyst. | |
As was confirmed by researches in recent years, POVs were considered to be fruitful for oxidative desulfurization due to their fast reversible multielectron transformation activity [31, 48-50]. However, catalytic reactions employing vanadium oxide clusters are rather rare, unlike those vanadium complexes and V-substituted POM-based compounds. Thus, the sulfur oxidation of diphenyl sulfide (DPS) were selected as benchmark system to evaluate the activity of compounds V6-1-4 in the sulfur oxidation reaction. Preliminarily, a series of contrastive explorations for oxidation of the model DPS by using V6–1 as the catalyst were performed to obtain optimum reaction conditions. Obviously, the best catalytic effect could be attained when using 10 mg V6–1, 4 mmol tert‑butyl hydroperoxide (TBHP) as the oxidant in 5 mL CH2Cl2 and at the temperature of 40 ℃ for 4 h (Fig. S21, Table S4 in Supporting information, entries 1–9). Under the aforementioned conditions, V6-2-4 also can catalyze the oxidation of DPS with commendable conversion (Fig. S23 in Supporting information), which shows better performance under even milder reaction conditions than some of the reported V-based heterogeneous catalysts (Table S5 in Supporting information). Additionally, as shown in Table S4, a quite low conversion was achieved once V6–1 was replaced by equimolar NH4VO3 or no TBHP existed in the reaction (Table S4, entries 11 and 13). Meanwhile, conversion only reaches 20% when the reaction was carried out without V6–1 (Table S4, entry 12). Therefore, we can conclude that both TBHP and V6–1 contribute to promoting desulfurization reaction efficiently. Consequently, the remaining catalytic experiments were performed with the presence of both V6–1 and TBHP in CH2Cl2 at 40 ℃ for 4 h
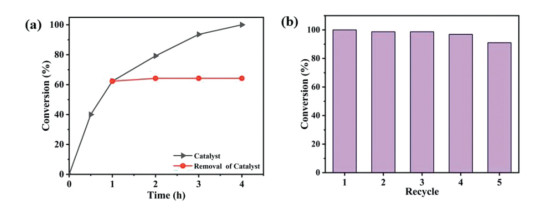
Excellent stability is crucial to catalytic applications. The superior stability of V6–1 is evidenced by PXRD patterns of V6–1 immersed in CH2Cl2 at different durations (Fig. S22 in Supporting information). Additionally, under the aforementioned conditions, we removed catalyst V6–1 from the reaction system when the reaction lasted for 1 h, subsequent conversion was not further boosted (Fig. 4a), illuminating that the reaction process is heterogeneous. Apart from good performance, another two important criteria for practical application, reusability and versatility, were also estimated. Thus, V6–1 was separated from the mixture by centrifugal filtration after each cycle, and the collected V6–1 was consecutively used in the next experiments. As depicted in Fig. 4b, the catalytic performance of V6–1 still retained well and V6–1 can be reused for at least four cycles with no significant decrease of conversion in the subsequent sulfoxidation reactions. Meanwhile, the infrared (IR) spectra (Fig. S24 in Supporting information) and PXRD patterns (Fig. S25 in Supporting information) of catalyst V6–1 after five circulations were in good agreement with those of the as-prepared samples, which manifested that V6–1 can maintain structural integrity after catalytic reaction.

|
Download:
|
| Fig. 4. (a) Catalytic dynamic (black) and hot filtration (red) studies for the sulfoxidation reactions by V6–1. (b) Recycling experiments for sulfoxidation reaction using V6–1 as the catalyst. | |
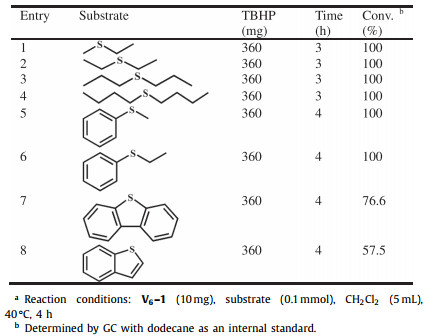
Finally, for the purpose of exploring the versatility of catalyst V6–1, a variety of alkyl and aryl sulfides were then selected as substrates to participate in sulfoxidation reactions under optimized conditions. And the corresponding results were provided in Table 2. The alkyl sulfides featuring with less steric hindrance were chosen as substrates, such as diethyl sulfide and dipropyl sulfide, satisfying conversion was reached with less time comparable to those of DPS (Table 2, entries 1–4). Regarding substituted derivatives of DPS (Table 2, entries 5 and 6), it is worth mentioning that the expected full conversions were obtained, when one benzene in DPS was replaced by a methyl or ethyl substituent with smaller steric hindrance. Subsequently, by reason of the challenge posed by larger steric hindrance of benzene rings in aryl thioethers, the dibenzothiophene and benzothiophene only affords the corresponding sulfoxide in 76.6% and 57.5% yield (Table 2, entries 7 and 8). That is to say, one main factor on desulfurization is identified to be the steric hindrance of sulfides, which shows good accordance with reported results [24, 29, 45].
|
|
Table 2 Results of other sulfide reactions catalyzed by V6–1 using TBHP as the oxidant.a |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The POM-based compounds have been broadly used for catalytic oxidative desulfurization [45, 51, 52]. As shown in Fig. S26 (Supporting information), it is proposed that V atoms initially interact with the oxidant TBHP to produce the active peroxovanadium complex. Then the O atoms from the peroxyvanadic acid complex attacks the sulfonium cation of the sulfur-containing substrate in a nucleophilic manner. As a result, the sulfides are oxidized to sulfone and sulfoxide.
In summary, a series of new hexanuclear vanadium oxide clusters modified by dual ligands, V6-1-4, have been successfully prepared under solvothermal conditions. All of them possess same principal structure {V6} cluster with Z-shaped configuration, which represents a brand-new hexanuclear vanadium cluster differing from other oxovanadium clusters in POV chemistry. Preliminary results indicated that V6–1 possess satisfactory catalytic activity toward CO2 cycloaddition reaction with epoxy compounds. Importantly, it can be recycled and reused at least three times with high catalytic activity. Expectantly, V6–1 as a heterogeneous catalyst was also found to be fruitful in the oxidation of sulfides. The triumphant isolation of the distinct {V6} cluster and decent catalytic effectiveness provide a feasible inspiration for the exploration of novel oxovanadium clusters and their catalytic property.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by Natural Science Foundation of Jiangsu (No. BK20191359), the Natural Science Foundation of China (No. 21571103) and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX22_1343).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.005.
| [1] |
J.D. Shakun, P.U. Clark, F. He, et al., Nature 484 (2012) 49-54. DOI:10.1038/nature10915 |
| [2] |
P. Nejat, F. Jomehzadeh, M.M. Taheri, et al., Renew. Sust. Energ. Rev. 43 (2015) 843-862. DOI:10.1016/j.rser.2014.11.066 |
| [3] |
B. Kirchner, B. Intemann, Nat. Chem. 8 (2016) 401-402. DOI:10.1038/nchem.2499 |
| [4] |
Z.Y. Du, Y.Z. Yu, N.F. Li, et al., Sustain. Energy Fuels 5 (2021) 3876-3883. DOI:10.1039/d1se00804h |
| [5] |
X. Yang, E.A. Fugate, Y. Mueanngern, L.R. Baker, ACS Catal. 7 (2016) 177-180. DOI:10.3390/ijgi5100177 |
| [6] |
R.C. Luo, Y.Y. Yang, K.C. Chen, et al., J. Mater. Chem. A 9 (2021) 20941-20956. DOI:10.1039/d1ta05428g |
| [7] |
R.C. Luo, X.Y. Liu, M. Chen, B.Y. Liu, Y.X. Fang, ChemSusChem 13 (2020) 3945-3966. DOI:10.1002/cssc.202001079 |
| [8] |
M. Reiter, S. Vagin, A. Kronast, C. Jandl, B. Rieger, Chem. Sci. 8 (2017) 1876-1882. DOI:10.1039/C6SC04477H |
| [9] |
R. Yamaguchi, C. Ikeda, Y. Takahashi, K.I. Fujita, J. Am. Chem. Soc. 131 (2009) 8410-8412. DOI:10.1021/ja9022623 |
| [10] |
P. Vidossich, A. Lledos, G. Ujaque, Acc. Chem. Res. 49 (2016) 1271-1278. DOI:10.1021/acs.accounts.6b00054 |
| [11] |
G.P. Yang, X.L. Zhang, Y.F. Liu, et al., Inorg. Chem. Front. 8 (2021) 4650-4656. DOI:10.1039/d1qi00485a |
| [12] |
G.P. Yang, K. Li, X.L. Lin, et al., Chin. J. Chem. 39 (2021) 3017-3022. DOI:10.1002/cjoc.202100397 |
| [13] |
Y.G. Dong, S. Chen, N.N. Jia, et al., Tungsten 3 (2021) 406-414. DOI:10.1007/s42864-021-00111-8 |
| [14] |
M. Aureliano, N.I. Gumerova, G. Sciortino, et al., Coord. Chem. Rev. 447 (2021) 214143-214156. DOI:10.1016/j.ccr.2021.214143 |
| [15] |
J.L. Wang, X.M. Liu, Z.Y. Du, Y. Xu, Dalton Trans. 50 (2021) 7871-7886. DOI:10.1039/d1dt00494h |
| [16] |
N. Li, J. Liu, B.X. Dong, Y.Q. Lan, Angew. Chem. Int. Ed. 59 (2020) 20779-20793. DOI:10.1002/anie.202008054 |
| [17] |
R.R. Langeslay, D.M. Kaphan, C.L. Marshall, et al., Chem. Rev. 119 (2018) 2128-2191. |
| [18] |
S. Chakraborty, B.E. Petel, E. Schreiber, E.M. Matson, Nanoscale Adv. 3 (2021) 1293-1318. DOI:10.1039/d0na00877j |
| [19] |
R.F. de Luis, J. Orive, E.S. Larrea, M.K. U.rtiaga, M.I. Arriortua, CrystEngComm 16 (2014) 10332-10366. DOI:10.1039/C4CE00532E |
| [20] |
Y.P. Zheng, Y. Tan, W.L. Zhou, et al., Inorg. Chem. 60 (2021) 12323-12330. DOI:10.1021/acs.inorgchem.1c01535 |
| [21] |
X. Yu, C.C. Zhao, J.X. Gu, et al., Inorg. Chem. 60 (2021) 7364-7371. DOI:10.1021/acs.inorgchem.1c00499 |
| [22] |
Y.Q. Gu, Q. Li, D.J. Zang, et al., Angew. Chem. Int. Ed. 60 (2021) 13310-13316. DOI:10.1002/anie.202011164 |
| [23] |
H.R. Tian, Z. Zhang, S.M. Liu, et al., J. Mater. Chem. A 8 (2020) 12398-12405. DOI:10.1039/d0ta00537a |
| [24] |
J.K. Li, C.P. Wei, Y.F. Han, et al., Dalton Trans. 50 (2021) 10082-10091. DOI:10.1039/d1dt01413g |
| [25] |
H.Y. An, J. Zhang, S.Z. Chang, Y.J. Hou, Q.S. Zhu, Inorg. Chem. 59 (2020) 10578-10590. DOI:10.1021/acs.inorgchem.0c00999 |
| [26] |
J.K. Li, X.Q. Huang, S. Yang, Y.Q. Xu, C.W. Hu, Cryst. Growth Des. 15 (2015) 1907-1914. DOI:10.1021/acs.cgd.5b00086 |
| [27] |
Y. Gu, Q. Li, D. Zang, et al., Angew. Chem. Int. Ed. 60 (2021) 13310-13316. DOI:10.1002/anie.202011164 |
| [28] |
X.Q. Huang, Y.N. Cui, J.H. Zhou, et al., Chin. Chem. Lett. 33 (2022) 2605-2610. DOI:10.1016/j.cclet.2021.09.042 |
| [29] |
H.M. Gan, C. Qin, L. Zhao, et al., Cryst. Growth Des. 21 (2021) 1028-1034. DOI:10.1021/acs.cgd.0c01375 |
| [30] |
J.P. Cao, Y.S. Xue, N.F. Li, et al., J. Am. Chem. Soc. 141 (2019) 19487-19497. DOI:10.1021/jacs.9b11146 |
| [31] |
K. Wang, Y.J. Niu, D.Y. Zhao, et al., Inorg. Chem. 56 (2017) 14053-14059. DOI:10.1021/acs.inorgchem.7b02207 |
| [32] |
B.K. Chen, X.Q. Huang, B. Wang, et al., Chem. Eur. J. 19 (2013) 4408-4413. DOI:10.1002/chem.201203854 |
| [33] |
Z.Y. Du, Y.Z. Yu, Y.L. Hong, et al., ACS Appl. Mater. Interfaces 12 (2020) 57174-57181. DOI:10.1021/acsami.0c18970 |
| [34] |
Q.D. Ping, J.P. Cao, Y.M. Han, et al., Inorg. Chim. Acta 517 (2021) 120198-120204. DOI:10.1016/j.ica.2020.120198 |
| [35] |
C. Daniel, H. Hartl, J. Am. Chem. Soc. 127 (2005) 13978-13987. DOI:10.1021/ja052902b |
| [36] |
J. Spandl, C. Daniel, I. Brvdgam, H. Hartl, Angew. Chem. Int. Ed. 115 (2003) 1195-1198. DOI:10.1002/ange.200390277 |
| [37] |
C. Aronica, G. Chastanet, E. Zueva, et al., J. Am. Chem. Soc. 130 (2008) 2365-2371. DOI:10.1021/ja078030q |
| [38] |
C. Daniel, H. Hartl, J. Am. Chem. Soc. 131 (2009) 5101-5114. DOI:10.1021/ja8073648 |
| [39] |
C.H. Ng, C.W. Lim, S.G. Teoh, et al., Inorg. Chem. 41 (2002) 2-3. DOI:10.1021/ic015574z |
| [40] |
Y. Gong, C.W. Hu, H. Li, J. Mol. Struct. 749 (2005) 31-35. DOI:10.1016/j.molstruc.2005.03.033 |
| [41] |
T. Kurata, A. Uehara, Y. Hayashi, K. Isobe, Inorg. Chem. 44 (2005) 2524-2530. DOI:10.1021/ic048751f |
| [42] |
J. Dong, P. Cui, P.F. Shi, P. Cheng, B. Zhao, J. Am. Chem. Soc. 137 (2015) 15988-15991. DOI:10.1021/jacs.5b10000 |
| [43] |
X. Wang, W.Y. Gao, Z. Niu, et al., Chem. Commun. 54 (2018) 1170-1173. DOI:10.1039/c7cc08844b |
| [44] |
Y.Q. Zhao, Y.Y. Liu, J.F. Ma, Cryst. Growth Des. 21 (2021) 1019-1027. DOI:10.1021/acs.cgd.0c01353 |
| [45] |
B.B. Lu, J. Yang, Y.Y. Liu, J.F. Ma, Inorg. Chem. 56 (2017) 11710-11720. DOI:10.1021/acs.inorgchem.7b01685 |
| [46] |
J. Liang, Y.B. Huang, R. Cao, Coord. Chem. Rev. 378 (2019) 32-65. DOI:10.1016/j.ccr.2017.11.013 |
| [47] |
J. Tang, F. Wei, S.J. Ding, et al., Chem. Eur. J. 27 (2021) 12890-12899. DOI:10.1002/chem.202102089 |
| [48] |
D. Liu, Y. Lu, H.Q. Tan, et al., Chem. Commun. 49 (2013) 3673-3675. DOI:10.1039/c3cc40990b |
| [49] |
J.K. Li, X.Q. Huang, S. Yang, et al., Inorg. Chem. 54 (2015) 1454-1461. DOI:10.1021/ic502447b |
| [50] |
R. Wan, P.P. He, Z. Liu, et al., Chem. Eur. J. 26 (2020) 8760-8766. DOI:10.1002/chem.201905741 |
| [51] |
J.W. Ding, R. Wang, Chin. Chem. Lett. 27 (2016) 655-658. DOI:10.7317/pk.2016.40.5.655 |
| [52] |
H.R. Tian, Z. Zhang, S.M. Liu, et al., Green Chem. 22 (2020) 7513-7520. DOI:10.1039/d0gc02812f |