 2023, Vol. 34
2023, Vol. 34 


b School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
Nitrogen-heterocycles are core frameworks of numerous naturally occurring products, alkaloids, drugs, and other biologically active compounds [1-3]. They also exist as key units in many functional materials [4] and, for example, have been intensively studied as basic materials in high-performance organic field-effect transistors (OFETs), organic light-emitting diodes (OLED), and organic photovoltaics (OPVs) [5, 6]. The versatile bio-activities and functionalities of nitrogen-heterocyclic compounds in various fields have initiated intensive efforts on the development of structurally diverse molecules and novel synthetic methods in modern organic synthesis [7-11]. Among those strategies discovered in the past decades, intermolecular cyclization between two or more substrates is the mostly used for preparing various highly functionalized nitrogen-heterocycles, with great progress in novel multi-component reactions (MCR) [12], facile metal-free systems [13-15], green aerobic oxidations [16-23], mild photoredox catalysis [24-30], and electrocatalytic transformations [31-37], etc.
On the other hand, ketones are fundamental compounds in chemical industry and academia. While ketones have been widely employed as raw materials in organic chemistry, their oxime derivatives, or ketoximes, have enabled much broader synthetic applications [38-43]. In the last decade, the ingenious internal oxidant strategy involving hydroxylamine derivative has emerged to feature excellent levels of chemo-selectivity for oxidative C—H functionalization [44-46]. Hence, C(sp2)–H activation with the ketoximes as an oxidizing directing group was achieved by the catalysis of Pd, Rh, Ru, Co, and among others, providing viable external-oxidant-free protocols for site-targeted C—H functionalization and nitrogen heterocycle synthesis. Furthermore, ketoximes bearing α-protons were generally employed as internal oxidant as well as C—C—N building blocks to enable condensation-based nitrogen-heterocycle formation [47, 48]. Within this field, our group focused on the development of novel catalytic systems to activate N—O bond of ketoximes to construct new nitrogen-heterocycle frameworks in the last few yields. With the principle of green chemistry [49-51], for example, we combined the internal oxidant strategy with aerobic oxidation and have realized the N—O bond activation and nitrogen-heterocycle formation under metal-free and even additive-free conditions. With respect to products obtained, beyond the formation of heterocycles by mono-annulation, the multi-component bis-annulations of ketoximes have been achieved for the first time, providing facile and straightforward entries to structurally valuable polycyclic heterocycles with highly extended π-conjugated systems. These findings have well complemented to the previous mainly copper-based catalytic systems and more importantly enriched the oxime chemistry in organic synthesis.
The current review is dedicated to showcase and discuss our recent progress in nitrogen-heterocycle formation from O-acyl ketoximes in the last few years, with an emphasis on the scope and limitations, as well as the mechanisms of these reactions. In these reactions, O-acyl ketoximes provide unique C—C—N building blocks for nitrogen-heterocycle formation. This review is organized according to the cyclization type such as formal [3 + 2] and [3 + 3] annulations, which led to different types of the five- and six-membered heterocyclic products, respectively. Another developed cyclization format is bis-annulation of ketoximes, which means the formation of polycyclic compounds with two new heterocycles within one reaction process.
2. Formal [3 + 2] annulationsThe formal [3 + 2] annulations of ketoximes with alkynes, alkenes, and imines have been previous developed [47]. These reactions generally proceeded under copper-catalyzed redox-neutral conditions. We suspected that oxidative systems would allow broader annulations of ketoximes with more coupling partners. Metal-free catalytic system rather than copper catalysis may be also competent to activate the N—O bond of ketoximes. Moreover, multi-component cascade [3 + 2] annulations of ketoximes had been still under developed.
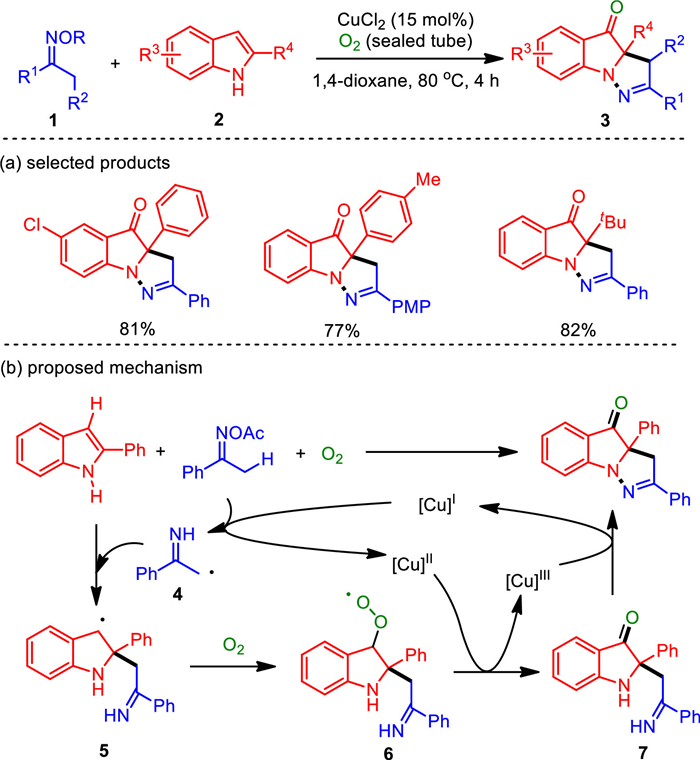
The introduction of functional groups on the indole ring is a hot research topic in synthetic chemistry. There are many catalytic systems for the mono- or di-functionalization of indoles, but the N-1, C-2 and C-3 tri-functionalization of indoles in one reaction is difficult to achieve [52-54]. In 2016, the aerobic oxidation and cyclization of indoles and ketoximes triggered by an internal oxidant and copper catalysis enabled N-1, C-2 and C-3 tri-functionalization of indoles in a one-pot manner [55]. It provided a way to obtain complex and pharmacologically significant pyrazo[1, 5-a]indole derivatives (Scheme 1). The co-oxidation conversion by internal oxidant under aerobic conditions also provided a new mode for dehydrogenation couplings. The optimized reaction conditions included 2-substituted indoles and ketoximes (1.2 equiv.) with 15 mol% of CuCl2 in 1, 4-dioxane (0.2 mol/L) under O2 atmosphere (sealed tube) at 80 ℃ for 4 h. This protocol showed good tolerance of a broad range of functionalities and formally released only water and acetic acid as the byproducts.

|
Download:
|
| Scheme 1. Aerobic oxygenation and cyclization of indoles under copper catalysis. | |
{kind=link}
Regarding the proposed reaction mechanism, the copper-initiated reductive N—O bond cleavage gives a methyl radical species 4 and [CuⅡ] species. The radical addition of 4 to indole double bond generates radical 5, which is trapped by dioxygen to furnish peroxy radical 6. The integration of 6 with [CuⅡ] followed by dehydroxylation affords a [CuⅢ] species and the imino intermediate 7, which could further coordinate with [CuⅢ] and deliver the final product through N—N bond reductive elimination.
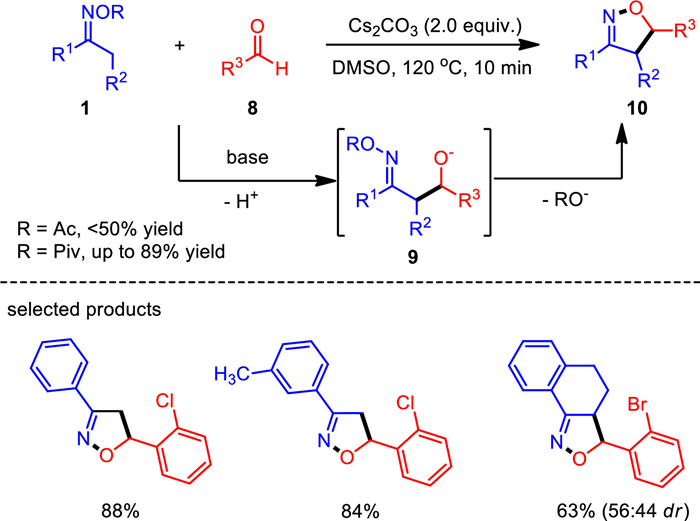
Isoxazoline has been widely discovered as a key unit in drugs and other biologically active compounds. Its derivatives are also used to construct β-amino acids, γ-amino alcohol and other functional molecule modules. Previous reports on hydroxylamine derivatives reveal that the N–O bond cleavage mainly rely on transition metal catalysts. In addition to copper-catalyzed N—O bond activation of ketoximes for heterocycle formation, a base-promoted [3 + 2] annulation of ketoximes and aldehydes has been developed for the synthesis of isoxazolines (Scheme 2) [56]. The key to the successful metal-free transformation was the pivalate leaving group of the oxime substrates, combined with cesium carbonate as promoter. This reaction allowed rapid assembly of a vast array of isoxazoline derivatives under simple base conditions. This method well complements to the classic isoxazoline formation via [3 + 2]-dipolar cycloaddition of nitrile oxides and olefins. When cyclic ketoxime esters were used as reactants, a series of isoxazoline fused heterocycles were generated, which could not be easily produced by previous methods. It represents a new reaction pattern involving electrophilic amination/N–O bond substitution.

|
Download:
|
| Scheme 2. Isoxazoline formation from ketoximes and aldehydes. | |
{kind=link}
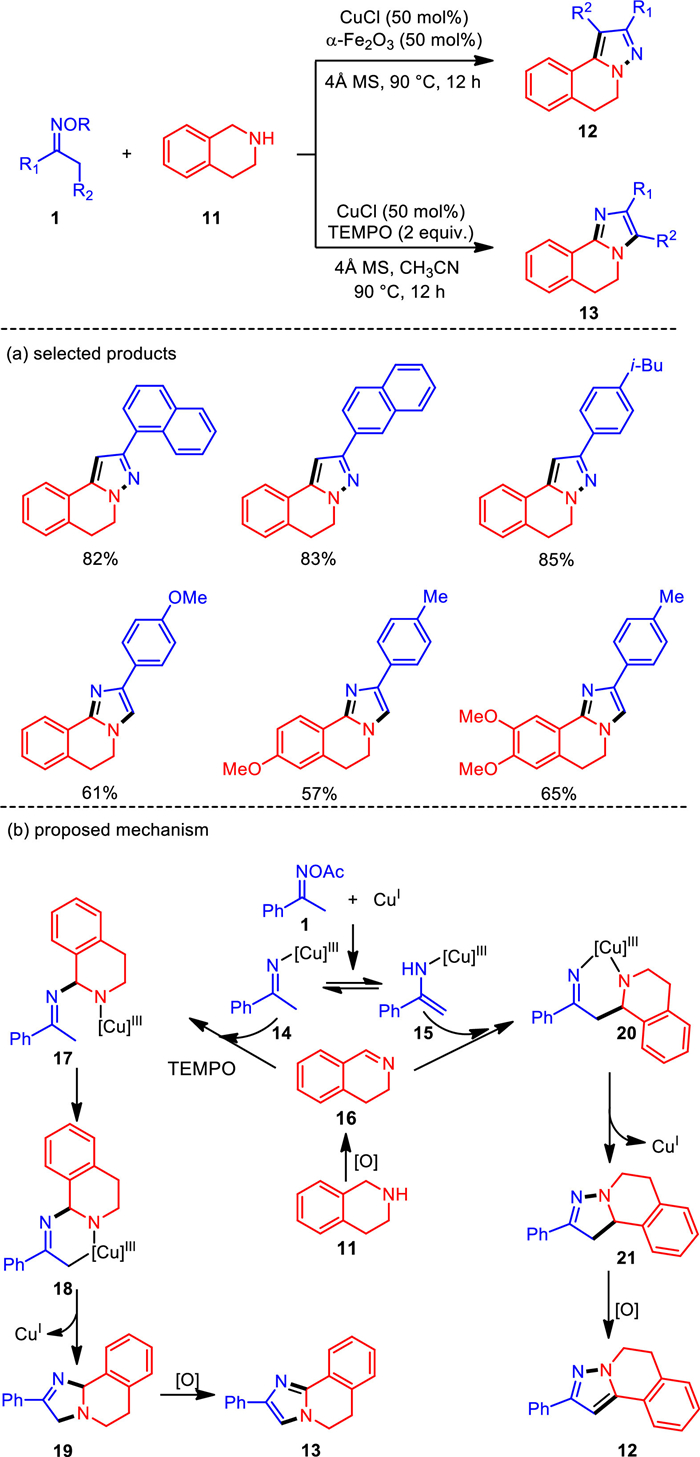
Methyl ketoximes were independently used as building blocks for the construction of pyrazoles [57, 58] and imidazoles [59, 60] with C—N double bonds of imines and pyridines via [3 + 2] formal annulations, respectively. The regioselectivity control in the annulations of oximes with the same kind C—N unsaturated bonds has not yet been achieved. Under altered oxidative systems, tetrahydroisoquinolines could be oxidized to the imine variants which coupled with ketoximes acetates in a regioselective manner under copper catalysis. With basically different oxidants, Fe2O3 and TEMPO, a series of structurally important pyrazole and imidazole products bearing isoquinoline moiety [61, 62] were selectively generated in good to excellent yields (Scheme 3) [63].

|
Download:
|
| Scheme 3. Regioselectivity control in the oxidative annulations of oximes with tetrahydroisoquinoline. | |
{kind=link}
Mechanistically, copper-mediated N—O cleavage of acetate oxime 1 leads to the imino Cu(Ⅲ) species 14, which can be tautomerized to enamino Cu(Ⅲ) intermediate 15. Simultaneously, tetrahydroisoquinoline is oxidized to dihydroisoquinoline 16. The nucleophilic migration insertion of 14 to 16 forms Cu(Ⅲ) intermediate 17. Then nucleophilic metallization of 17 gives the six-membered copper ring intermediate 18. The imidazole product 13 is obtained by dehydogenation after reductive elimination of 18. Alternatively, the nucleophilic addition of enamine 15 to dihydroisoquinoline 16 affords Cu(Ⅲ) species 20, which undergoes N—N bond reductive elimination to generate dihydropyrazole 21. Then, oxidative dehydrogenative aromatization occurs to produce pyrazole product 12. The key to excellent regioselectivities should be the tautomerization of imino and enamino Cu(Ⅲ), which is probably altered by different oxidants.
The oxazole skeleton is an important N-heterocyclic building block. There are many ways to synthesize polysubstituted oxazoles [64], but few of them could introduce trifluoromethyl (−CF3) group on the oxazole rings, which may change biological activities of the oxazole compounds. Therefore, the development of simple and efficient preparation methods of trifluoromethyl-modified oxazoles is a research hotspot in molecular synthesis. In 2019, the copper-catalyzed three-component oxidative annulation reactions of oxime, trifluoroacetic anhydride and arylthiols were developed to prepare 2-trifluoromethyl oxazoles (Scheme 4) [65]. This transformation combines N—O bond cleavage, C—H functionalization and intramolecular annulations, and provides an effective route to diversely trisubstituted oxazole products in good yields. Salient features of this protocol include good tolerance to various functional groups such as alkyl, ester and nitro groups.

|
Download:
|
| Scheme 4. Synthesis of 2-trifluoromethyl oxazoles. | |
{kind=link}
In terms of reaction mechanism, first, p-toluenethiol 22 is oxidized by NIS to generate sulfur radical intermediate 25. And the oxime trifluoroacetate 26 is generated in situ from the acylation of the ketoxime. The treatment of 26 by Cu(Ⅰ) forms the imino radical 27 through single electron transfer (SET). Acylation and isomerisation of 27 give alkyl radical intermediate 28, which is coordinated with Cu(Ⅰ) and sulfur radical 25 to generate alkyl Cu(Ⅲ) intermediate 29. The reductive elimination occurs to generate imine 30, with following isomerization to produce the enamine intermediate 31. Finally, the oxazole product is generated by oxidative cyclization through radical oxidation process of intermediates 32–34.
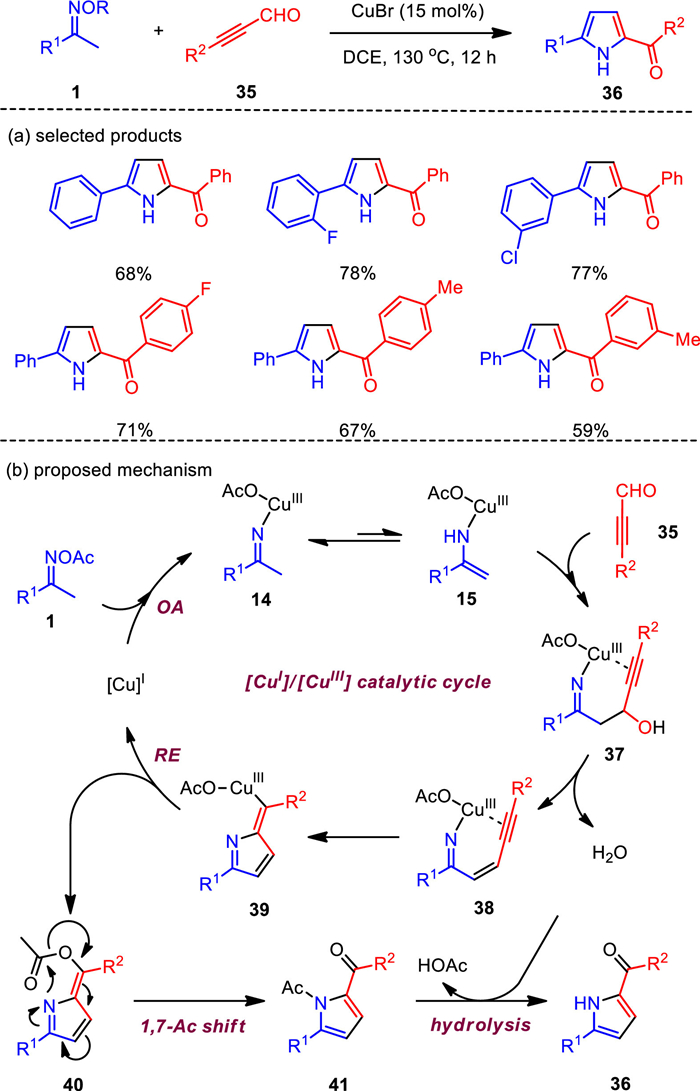
The C—C—N fragments of O-acyl ketoximes were used to react with a series of unsaturated compounds to obtain various pyrrole derivatives [66-71]. Although these methods are practical, they limit the types of pyrrole products, and the structurally important 2-acylpyrroles has not yet been accessed by this strategy [72]. To this end, propynal compounds were employed as the coupling partner for the first time, which smoothly reacted with methylketoximes under copper-catalyzed [3 + 2] formal annulations (Scheme 5). Notably, the chemo-selective formation of two 2-acylpyrroles was achieved to provide an unique way for valuable 2, 5-disubstituted and 1, 2, 5-trisubstituted pyrrole compounds [73]. The cyclization reaction has high regioselectivity and chemoselectivity, which well complements the previous annulations, especially the formation of pyrroles. More importantly, this reaction system also explored the three-component assembly of pyrrole by simply adding carboxylic acid.

|
Download:
|
| Scheme 5. Synthesis of acylpyrroles through Cu-catalyzed cascade cyclization of ketoximes. | |
{kind=link}
The reaction mechanism was initially proposed on the basis of some control experiments. First, copper(Ⅰ) is oxidatively added (OA) to the N—O bond of the oxime group to obtain imino copper(Ⅲ) species 14, and 14 is tautomerized to obtain enamino copper complex 15. Subsequently, the nucleophilic 1, 2-addition of 15 to alkynal 35 affords intermediate 37, which undergoes dehydration reaction to generate enyne imine copper(Ⅲ) complex 38. Then the intramolecular migration insertion of N—Cu bond into the C—C triple bond generates intermediate 39, which is transferred to 2-methylene-2H-pyrrole 40 and copper(Ⅰ) through reduction elimination (RE), completing the copper catalytic cycle. Finally, the N-acylpyrrole product 41 is furnished through aromatization tautomerization/acyl shift, and then hydrolyzed to give 2, 5-disubstituted pyrrole 36. It is worth noting that the [CuⅠ]/[CuⅡ] catalytic cycle is also suitable for ketoxime N—O bond activation.
3. Formal [3 + 3] annulationsDue to the importance of polysubstituted pyridines in industrial applications, the synthesis of polysubstituted pyridines from acyclic precursors has always been a hot spot in synthetic chemistry [74-78]. The unique C—C—N building blocks from ketoximes had been successfully employed to construct pyridine motif through formal [3 + 3] annulations with C3 units such as acroleins and allylic alcohols [47]. Again, copper catalysis was indispensible in these reactions. Furthermore, while previous directing C—H activation/annulations of aromatic ketoximes with alkynes efficiently enabled the formation of fused pyridines through internal oxidant strategy [44], we speculated that the C—C—N unit of methylketoximes would be also exploited with aromatic aldehydes to construct fused pyridine compounds, such as quinolines, via formal [3 + 3] annulations.
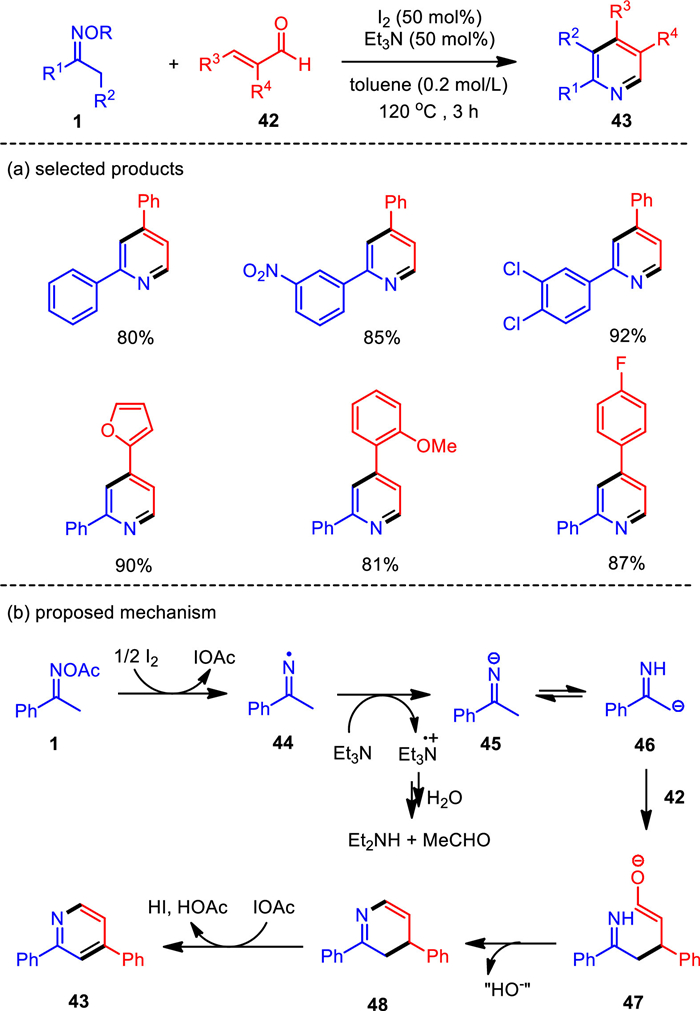
In complementary to previous copper-catalyzed pyridine formation from ketoximes and α, β-unsaturated aldehydes (42) [79-81], we have proved for the first time that the metal-free catalytic system with the combination of iodine and triethylamine could effectively trigger the oxime-based pyridine synthesis (Scheme 6). This method provided a facile access to 2, 4-disubstituted, 2, 3, 4-trisubstituted, and 2, 3, 4, 5-tetrasubstituted pyridine products with high yields, high chemical selectivity, and wide functional group tolerance [82]. In this system, iodine was demonstrated to be highly effective for the 1e− reduction of oximes, generating an imino radical which reacted with cinnamaldehyde with high efficiency to give functionalized pyridines. The combine of iodine and triethylamine had been proved to be effective in activating the N—O bond of oximes, and thus enabling the assembly of pyridines. It opened a new point for the synthesis of oxime-based N-heterocycle in metal-free systems.

|
Download:
|
| Scheme 6. Iodine-mediated pyridine formation. | |
{kind=link}
Based on the experimental observation, a plausible reaction mechanism was proposed for the metal-free hetero-cyclization of oximes and enals. As shown in Scheme 6b, the I2-mediated reduction of oxime 1 gives a [I+] species and imino-radical 44, which traps an electron to form the corresponding anion 45. Then Michael-type addition of the carbon-centered anion 46, generated by 1, 3-H-shift of 45, would take place with the unsaturated aldehydes to generate intermediate 47, which eliminates hydroxyl anion to furnish the dihydropyridine 48. Finally, the desired pyridine 43 is afforded through aromatization of 48 with the [I+] species as the main oxidant. Additionally, one molecular Et3N as the base could provide two electrons and decays itself into Et2N+ = CHCH3, which would be further hydrolyzed into secondary amine and aldehyde. It is therefore reasonable to utilize 0.5 equiv. of Et3N as the electron donor.
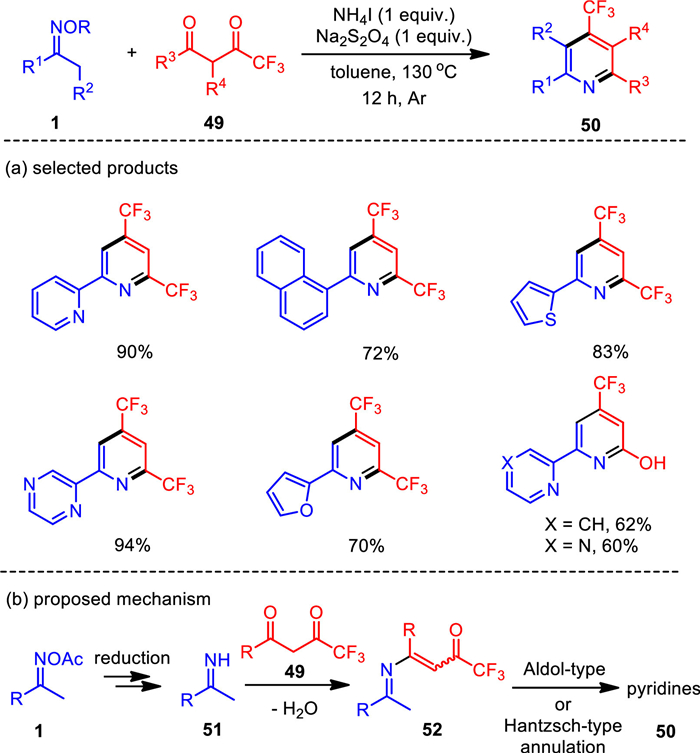
Due to the successful applications of organic fluoro molecules in pharmaceutical research, synthetic fluorine chemistry set off a wave of research fever. In 2017, a reductive condensation reaction of ketoximes and trifluoromethyl 1, 3-diketones was realized under metal-free conditions (Scheme 7) [83]. The combined NH4I/Na2S2O4 reductive system was found to be effective to facilitate oxime N—O bond cleavage and the resultant C—C—N building blocks successfully coupled with 1, 3-diketones to generate structurally valuable trifluoromethyl pyridines with good to excellent yields. The robust nature of this reaction was reflected by the broad tolerance of functionalities and effective gram-scale synthesis with satisfactory yield. The present NH4I/Na2S2O4 system was also expanded to the three-component assembly of trifluoroacetyl pyridines with the addition of aldehydes through Hantzsch-type cyclization.

|
Download:
|
| Scheme 7. Synthesis of fluorinated pyridines. | |
{kind=link}
Unexpected results were observed when we utilized trifluoromethylethylacetoacetate as the coupling partner, which afforded pyridin-2-ols as the sole product in moderated yields. In this procedure, ethanol instead of H2O was eliminated in the condensation step. Therefore, this protocol also provides a convenient entry to the 4-trifluoromethyl-pyridin-2-ol motif.
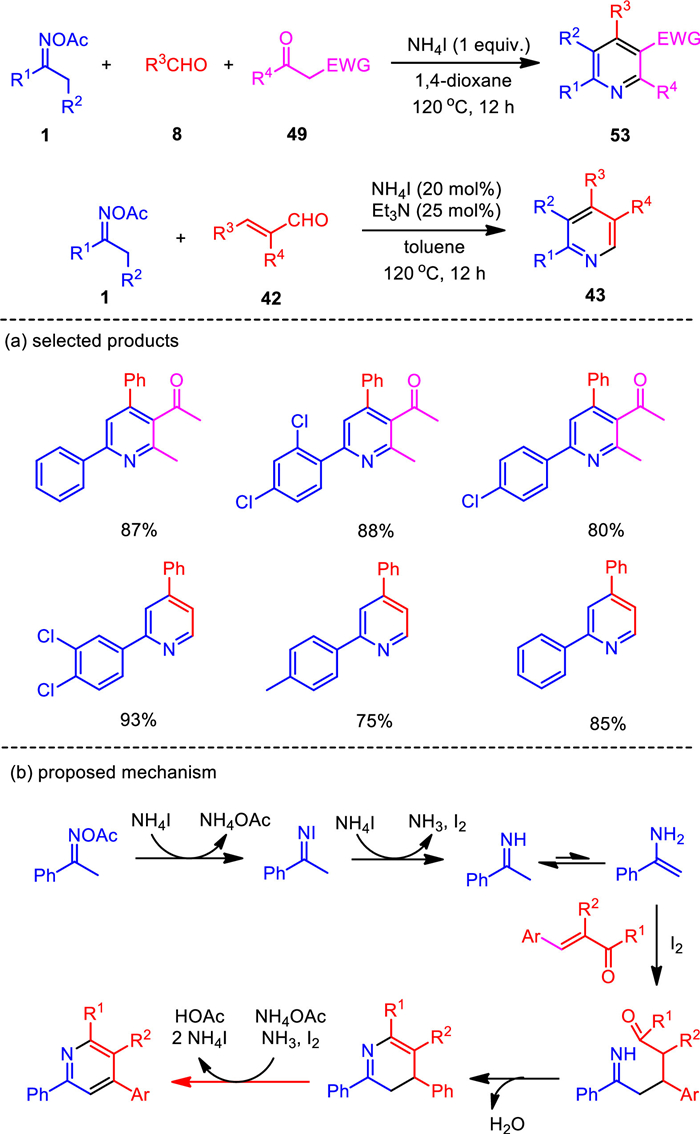
The NH4I-based catalytic system for oxime N—O bond activation was found to be a more general platform for pyridine synthesis. It was also smoothly applied to the formal [3 + 3] annulations of ketoximes and acroleins, as well as the three-component reactions of ketoximes, benzaldehydes, and 1, 3-diketones (Scheme 8) [84]. While the three-component reaction of oxime acetates, benzaldehydes, and 1, 3-dicarbonyls proceeded well by the assistance of stoichiometric amount of ammonium iodide, the condensation of oximes and acroleins was enabled by catalytic initiator to afford substituted pyridines. By this protocol, substituted pyridine products were generated in moderate to excellent yields with a broad range of functional groups tolerated. Compared with the iodine-promoted oxime reduction probably involving a radical pathway, ammonium iodide may directly serve as both electron and proton donors to reduce the N–O bond of oxime acetate to give imine and acetic acid, in which elemental iodine would be generated, while radical intermediate would not be formed.

|
Download:
|
| Scheme 8. NH4I-mediated pyridine formation. | |
{kind=link}
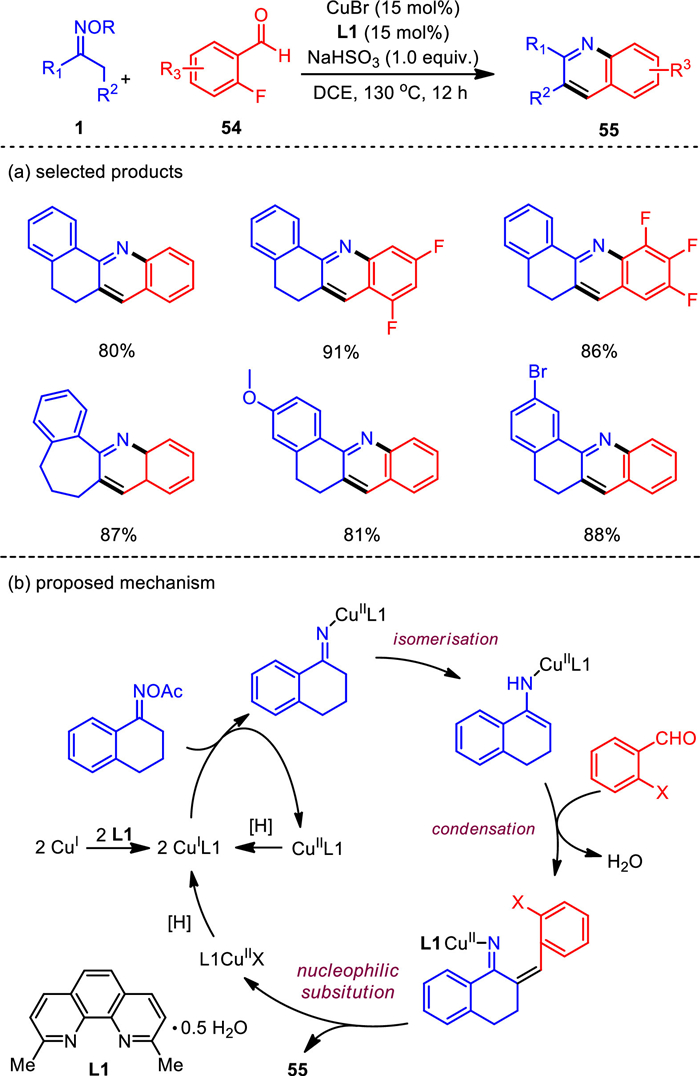
Due to the structural importance of quinoline motif in pharmaceuticals, there is an urgent need for concise and practical methods to obtain functionalized quinoline compounds. Inspired by the pyridine construction using C—C—N building blocks of ketoximes, we dedicated for a long time to exploit aromatic coupling partners to achieve quinoline formation through [3 + 3] annulation with ketone oximes. As a structural variant of acroleins, benzaldehyde may be directly used, but after a serious of attempts we failed to obtain any corresponding cyclization products from it. Then, we found that the ortho functionalized benzaldehydes with halo, methoxy, and nitro groups were all capable in such a [3 + 3] annulation reaction. Of them, o-fluorobenzaldehydes resulted in the best yield. Moreover, high conversion was observed when NaHSO3 or Na2S2O4 was employed as the additive, which may serve as a reducing reagent to activate oximes [85]. Other optimal parameters significantly facilitating the reaction included the use of CuBr as the catalyst and 2, 9-dimethyl-1, 10-phenanthroline as the ligand. This novel copper-based catalytic system offers an efficient method for the synthesis of substituted quinoline derivatives with overall satisfactory yields and a broad range of compatible functionalities (Scheme 9). Furthermore, with simple treatment of the resultant products, this work also provides a rapid access to synthetically and pharmaceutically useful quinoline-fused polycycles such as benzo[c]acridines.

|
Download:
|
| Scheme 9. Copper-catalyzed formal [3 + 3] annulations of ketone oximes and o-fluorobenzaldehydes. | |
{kind=link}
4. Bis-heteroannulations
The transition metal-catalyzed C(sp2)–H activation with the N-oxyenamine moiety as an oxidizing directing group was widely explored in the last few decades, providing viable external-oxidant-free protocols for site-targeted C(sp2)–H functionalization and heterocycle synthesis. On the other hand, ketoximes bearing α-protons were generally employed as internal oxidant as well as C—C—N building blocks to enable condensation-based N-heterocycle formation. Despite tremendous advances in thiazole synthesis by this viable strategy [86-88], the construction of highly functionalized molecules under facile reaction conditions has been still underdeveloped. We supposed that the ortho C(sp2)–H functionalization and N-heterocycle formation using the C—C—N unit of acetophenone oximes would be concurrently realized within one reaction process. This bis-annulation strategy seemed to be very attractive because it could highly improve the complexity of resultant products from simple raw materials.
Thiophene-fused polycyclic π-conjugated systems have broad application prospects in the synthesis of high-performance optoelectronic materials [89-91]. In 2018, we reported the first two-fold heteroannulation of oxime internal oxidants initiated by the copper-catalyzed activation of N—O, with subsequent cascade formation of multiple C–S bonds [92]. This three-component protocol starts from easily available ketoximes, aldehydes, and elemental sulfur and provides rapid access to thiophene-fused polycyclic heteroarene products with high atom economy (Scheme 10). The CuBr/Li2CO3 combined catalytic system is quite simple, while enables efficient construction of complex molecules. Both aromatic and vinyl ketoximes were compatible with this system and a diverse range of thieno[3, 2-d]thiazole and benzothieno[3, 2-d]thiazole products were synthesized with generally moderate to excellent yields. Mechanistic studies revealed that the copper catalyst enabled the activation of oxime N–O bond to form a highly active imino radical, which induced the cascade bis-heteroannulation reaction. Moreover, 3-aminobenzothiophene provided products in good yields with or without the assistance of copper catalysts. These results indicate that the formation of the thiazole ring is the final process, and the copper catalyst does not function in the formation of thiazole ring. Control experiments also showed dithiazole product (60) was produced in the absence of aldehydes, suggesting the formation of polysulfide imine intermediate (59) by radical incorporation of elemental sulfur to activated oximes.

|
Download:
|
| Scheme 10. Three-component bis-heteroannulations of ketoximes, aldehydes, and elemental sulfur. | |
{kind=link}
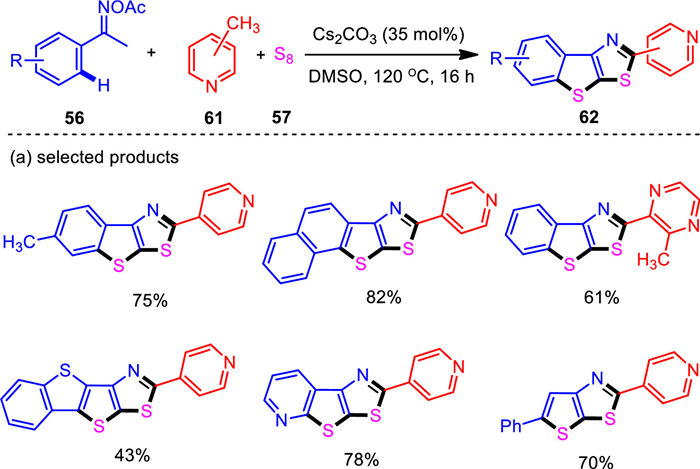
One of the important strategies for the synthesis of complex organic compounds is the direct functionalization of unactivated methylheteroarene C—H bonds. With the established bis-annulations of ketoximes with aldehydes as the one carbon source, methylheteroarenes were successfully used instead of aldehydes [93]. Control experiments indicated that copper catalyst was not required. Thus, a base-promoted catalyst-free system for the sequential self-assembly of fused thieno[3, 2-d]thiazoles was developed (Scheme 11). In this system, elemental sulfur was proven to play tri-function roles to activate other two components and as an efficient user-friendly sulfur source. The present protocol provides a viable access to a diverse range of functionalized thieno[3, 2-d]thiazoles with a variety of synthetically useful functionalities accommodated. Besides methylpyridines, other effective methyl N-heteroarenes including pyrazine, quinoxaline, quinoline and isoquinoline moieties, furnished the corresponding products in generally moderate yields. The robust nature of the present reaction was reflected by the effective gram-scale experiment, in which the product of model reaction was given in a satisfactory yield. The resultant thieno-fused poly-heterocyclic product was fluorescent under ultraviolet light and the studies on photophysical properties revealed the potential of this compound as a fluorescent indicator for intracellular pH sensing. Mechanistic studies revealed that the cleavage of the pyridinyl methyl C—H bonds was the rate-determining step of this reaction.

|
Download:
|
| Scheme 11. Three-component bis-heteroannulations of ketoximes, methylheteroarenes, and elemental sulfur. | |
{kind=link}
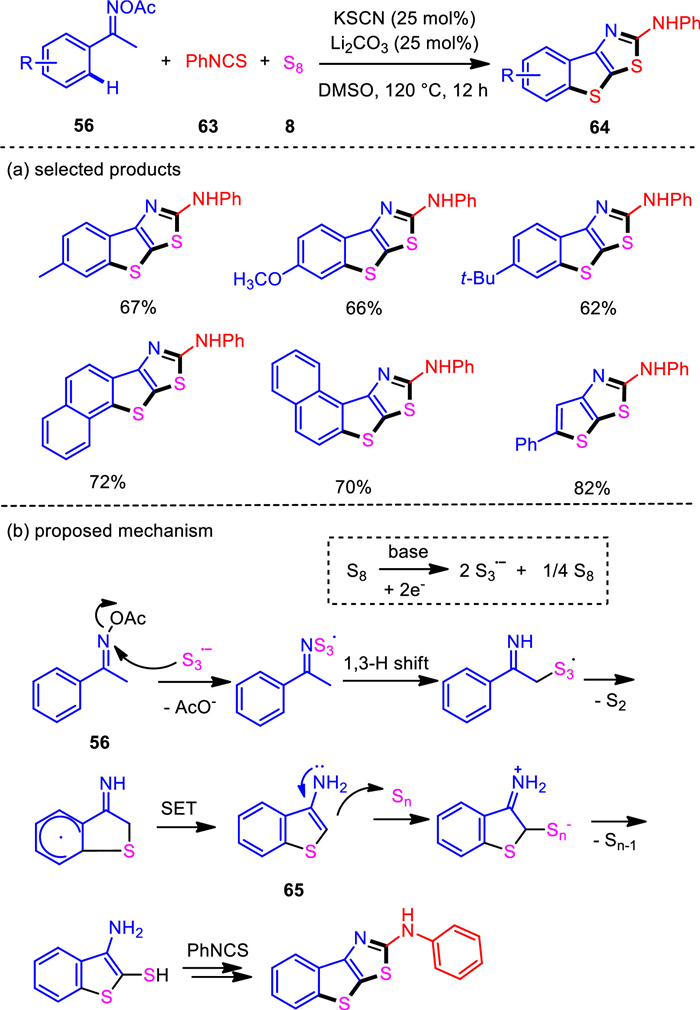
While the use of isothiocyanates as the coupling partners with ketoximes afforded 2-aminothiazoles, which was disclosed by Jiang group [87], we discovered the three-component bis-heteroannulation of methylketoxime esters, isothiocyanates, elemental sulfur by synergistic base system to produce 2-aminobenzo[4, 5]thieno[3, 2-d]thiazoles with high selectivity (Scheme 12) [94]. This protocol provided a unique way to obtain amino-modified products [95] through copper-free bis-thienoannulation. Mechanism studies indicated that under the alkaline conditions, the generated tri-sulfur radical anion [S3]•− would be the key initiator to activate acetic oximes. In the present system, 3-aminobenzothiophene (65) was probably delivered by radical sulfuration and cyclization. Control experiments showed that 3-aminobenzothiophene did not lead to the desired product in the absence of elemental sulfur, indicating a three-component cyclization in the process of thiazole ring formation.

|
Download:
|
| Scheme 12. Three-component bis-heteroannulations of ketoximes, isothiocyanates, and elemental sulfur. | |
{kind=link}
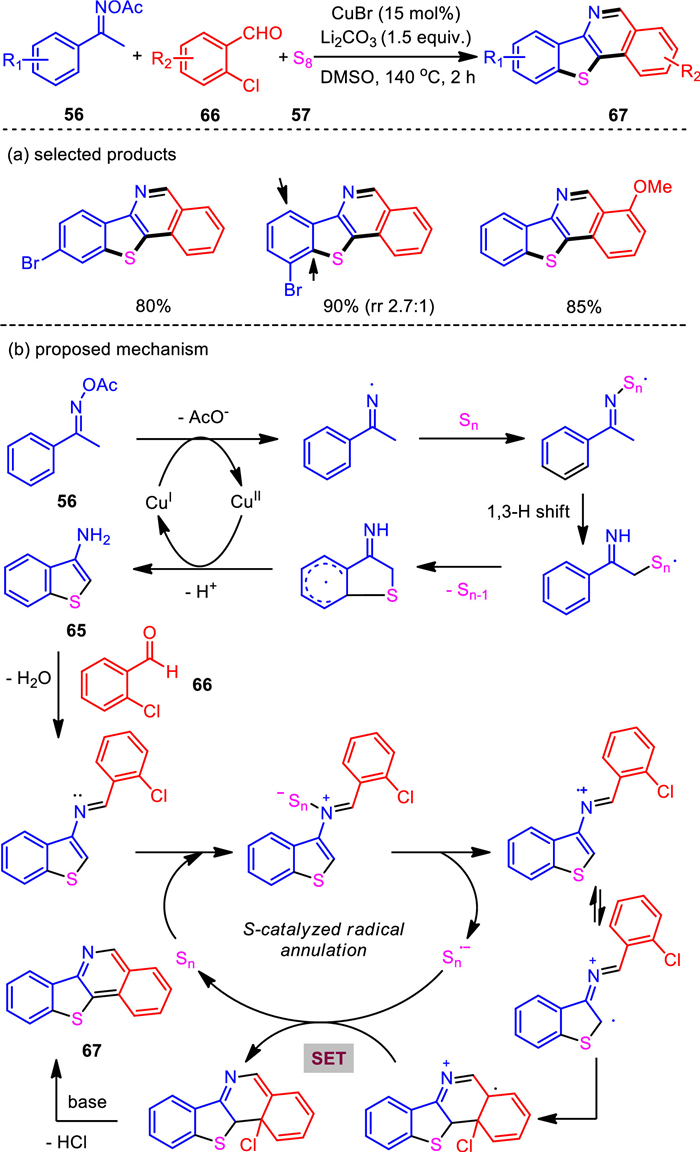
When we subjected o-bromobenzaldehyde as the coupling partner in the bis-heteroannulation reaction under the CuBr/Li2CO3 system, besides the formation of the desired benzo[4, 5]thieno[3, 2-d]thiazole, a benzo[4, 5]thieno[3, 2-c]isoquinoline product was isolated. Due the structural complexity of benzo[4, 5]thieno[3, 2-c]isoquinoline and thieno[3, 2-c]isoquinoline compounds, reports on the construction of this motif are rare. It provides a direct method for the synthesis of thiophene-fused polycyclic π-conjugated systems. Hence, we optimized the conditions of this new bis-heteroannulation reaction. Unexpectedly, o-chlorobenzaldehyde was found to be superior to o-bromobenzaldehyde for this reaction, exclusively affording the benzo[4, 5]thieno[3, 2-c]isoquinoline product. Increased amount of base loading and elevated reaction temperature were both beneficial to enhance the desired transformation. Under the optimized reaction conditions, various benzo[4, 5]thieno[3, 2-c]isoquinoline and thieno[3, 2-c]isoquinoline compounds were accessed with generally good yields even in the gram-scale preparation. Mechanistic studies revealed a two-step process involving a sequential copper and sulfur catalysis relay (Scheme 13) [96].

|
Download:
|
| Scheme 13. Three-component bis-heteroannulations of ketoximes, o-chlorobenzaldehydes, and elemental sulfur. | |
{kind=link}
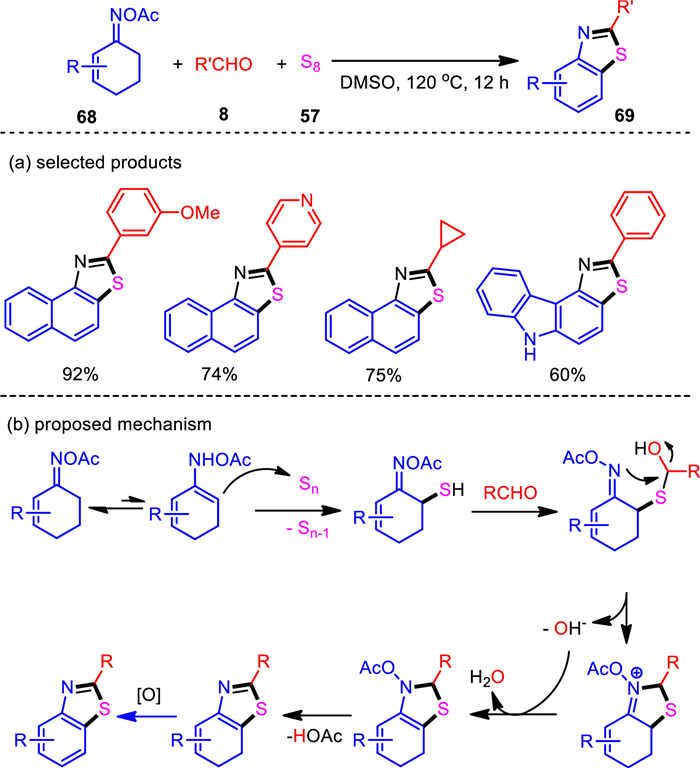
Catalytic dehydrogenative aromatization of saturated cyclic hydrocarbons is an effective method for molecular synthesis [97, 98], where various aromatic compounds can be obtained through dehydrogenation aromatization. Within our continue interest in heterocycle formation from ketoximes, the three-component cascade dehydrogenative aromatization and cyclization process was explored. The oxime acetates derived from 1-tetralones and cyclohexenones were employed as the precursors, which successfully coupled with benzaldehydes and elemental sulfur under catalyst- and additive-free conditions (Scheme 14) [99]. Thus, the new benzothiazole ring system was constructed in a sustainable one-pot manner. The resultant naphtho[1, 2-d]thiazoles and benzo[d]thiazoles were obtained with a wide range of compatible functionalities. Notably, heteroaryl and aliphatic aldehydes also featured good reactivities. Heteroaryl-fused cyclohexenone oximes were found to work well, leading to an unique and efficient access to benzofuro[4, 5-d]thiazole, benzothieno[4, 5-d]thiazole, and thiazolo[5, 4-c]carbazole motifs. Since 3, 5-diarylcyclohexenones could be conveniently prepared through a reaction sequence with Aldol condensation followed by Robinson annulation, the present protocol provides a stepwise modular assembly of 2, 5, 7-triarylbenzo[d]thiazoles starting from six easily available components including acetophenone, two aldehydes, acetone, hydroxylamine, and elemental sulfur. Furthermore, all the procedures involved no transition metals and other environmentally undesired reagents. Mechanistically, the reaction proceeded via a sequential cascade of Willgerodt-kindler-type sulfuration, thiazoannulation, and dehydrogenative aromatization process.

|
Download:
|
| Scheme 14. Catalyst- and additive-free annulation/aromatization leading to benzothiazoles and naphthothiazoles. | |
{kind=link}
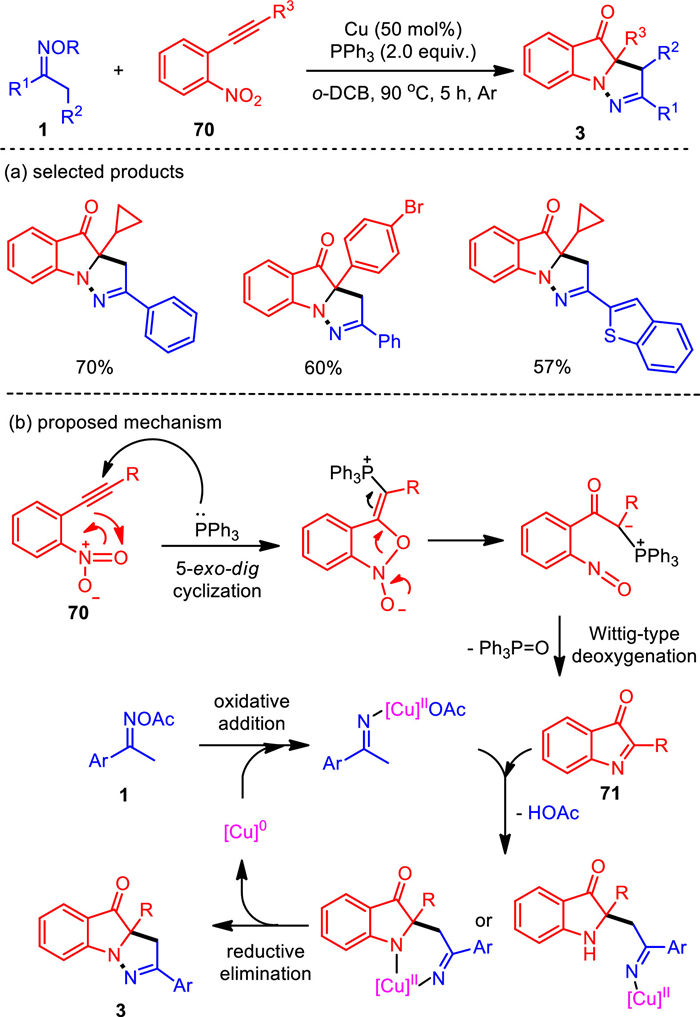
Inspired by our aerobic oxidation and cyclization of indoles with ketoximes, we designed the reductive bis-annulation reaction of o-nitroalkynes with ketoximes. The synergistic copper(0)/PPh3 system was found to be efficient for this cascade reaction (Scheme 15) [100]. It also conquered the key challenge of the formal conflict between the initial reductive cycloisomerization and the following oxidative N—N bond generation in one pot of this domino transformation. This protocol provides a complementary access to a diverse range of structurally and pharmacologically significant pyrazo-fused pseudoindoxyl compounds. Synthetically useful functional groups, including sensitive C—I bond, are compatible with this system. Mechanistic studies suggest a different pathway from previous work. Therein, the PPh3-trigered reductive cycloisomerization reaction resulted in the key indol-3-one intermediate (71), which subsequently coupled with methylketoximes to generate the dihydropyrazole ring under copper mediation.

|
Download:
|
| Scheme 15. Synthesis of pseudoindoxyls from ketoximes and o-nitroalkynes. | |
{kind=link}
5. Conclusions
Owning to the advantages of sustainable chemistry, enormous efforts have been dedicated to the development of green technologies and sustainable chemical processes. The use of O-acyl ketoximes as an efficient C—C—N building block for heterocycle formation represents a facile and versatile strategy, due to the embodied features including generally high atom economy and the structural diversity of resultant nitrogen-containing heterocycles. The novel ring systems constructed by our group include pyrazo[1, 5-a]indole, isoxazoline, pyrazolo[5, 1-a]isoquinoline, imidazo[2, 1-a]isoquinoline, trifluoromethylpyridine, trifluoromethyloxazole, quinoline, thieno[3, 2-d]thiazole, benzo[4, 5]thieno[3, 2-c]isoquinoline, and benzo[d]thiazole, and so forth. Furthermore, the discovery of these diverse catalytic systems will open new entries to broader applications to synthesize structurally complex and valuable functionalized molecules.
With respect to catalytic systems, a range of copper catalysis and base-promoted systems have been developed. Mechanistically, copper-mediated 1e− reduction of the N–O σ bond forms highly active imino radical or otherwise oxidative addition of low-valance copper(Ⅰ) to N–O σ bond to give imino-metal complexes. Specifically, copper-catalyzed oxidative reactions generally involve the formation of copper(Ⅲ) species and imino radicals, and the activation the N—O bond of oximes by catalysts could also form the NH imine in the process with reductive reagents. Moreover, the use of radical initiator, such as elemental iodine and sulfur, would also lead to the formation of imino radical intermediates. While the resultant NH imine intermediate generally proceeds through condensation reaction with unsaturated hydrocarbon, carbonyls, or other coupling partners, the highly active imino radical would trigger cascade radical couplings and cyclization. In this regard, much more novel reactants can be designed to trap the active intermediates to discover more reactivities of ketoximes.
In spite of significant utilities, these newly developed systems of heterocycle formation from ketoximes still suffer from limitations. The ketoximes used in our systems provide C—C—N building blocks to convergently form five- or six-membered nitrogen heterocycles. Other type of products such as medium size heterocycles are hardly accessed. Although mild photoredox catalysis could be used to activate certain weak N—O bond of oximes [47], thermal conditions (> 80 ℃) are generally required in our system with simple acetoxy as leaving group. Hence, it is still demand to further develop the versatility and expand the feasibility of oxime chemistry. In the last few years, sustainable electrocatalysis and visible light-induced photocatalysis have emerged to be versatile tools for organic synthetic chemistry. These popular techniques may have more applications in the transformations of ketoximes for heterocycle formation in the near future.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support by the National Natural Science Foundation of China (No. 22071211), the Science and Technology Planning Project of Hunan Province (No. 2019RS2039), Hunan Provincial Natural Science Foundation of China (No. 2020JJ3032), Scientific Research Fund of Education Department of Hunan Province (No. 21A0079), and Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University (No. 2022C02) is gratefully acknowledged.
| [1] |
H.H. Nguyen, M.B. Kim, R.J. Wilson, et al., J. Med. Chem. 61 (2018) 7168-7188. DOI:10.1021/acs.jmedchem.8b00450 |
| [2] |
H.D. Showalter, J. Nat. Prod. 76 (2013) 455-467. DOI:10.1021/np300753z |
| [3] |
J.A. Homer, J. Sperry, J. Nat. Prod. 80 (2017) 2178-2187. DOI:10.1021/acs.jnatprod.7b00390 |
| [4] |
I.A. Stepek, K. Itami, ACS Mater. Lett. 2 (2020) 951-974. DOI:10.1021/acsmaterialslett.0c00206 |
| [5] |
R.D. McCullough, Adv. Mater. 10 (1998) 93-116. DOI:10.1002/(SICI)1521-4095(199801)10:2<93::AID-ADMA93>3.0.CO;2-F |
| [6] |
D.T. McQuade, A.E. Pullen, T.M. Swager, Chem. Rev. 100 (2000) 2537-2574. DOI:10.1021/cr9801014 |
| [7] |
X.F. Wu, H. Neumann, M. Beller, Chem. Rev. 113 (2013) 1-35. DOI:10.1021/cr300100s |
| [8] |
S. Tian, T. Luo, Y. Zhu, et al., Chin. Chem. Lett. 31 (2020) 3073-3082. DOI:10.1016/j.cclet.2020.07.042 |
| [9] |
Y. Wu, Y.W. Lin, W.M. He, Chin. Chem. Lett. 31 (2020) 2999-3000. DOI:10.1016/j.cclet.2020.09.005 |
| [10] |
S. Peng, Y. Lin, W. He, Chin. J. Org. Chem. 40 (2020) 541-542. DOI:10.6023/cjoc202000006 |
| [11] |
S. Han, L. Hu, Y. Zhang, et al., Chin. J. Org. Chem. 41 (2021) 1053-1071. DOI:10.6023/cjoc202007036 |
| [12] |
V. Estevez, M. Villacampa, J.C. Menendez, Chem. Soc. Rev. 43 (2014) 4633-4657. DOI:10.1039/C3CS60015G |
| [13] |
J. Lv, B. Zhao, Y. Han, et al., Chin. Chem. Lett. 32 (2021) 691-694. DOI:10.1016/j.cclet.2020.06.028 |
| [14] |
X.M. Xu, D.M. Chen, Z.L. Wang, Chin. Chem. Lett. 31 (2020) 49-57. DOI:10.1016/j.cclet.2019.05.048 |
| [15] |
H. Zhuang, H. Li, S. Zhang, et al., Chin. Chem. Lett. 31 (2020) 39-48. DOI:10.1016/j.cclet.2019.06.027 |
| [16] |
X. Ji, D. Li, X. Zhou, et al., Green Chem. 19 (2017) 619-622. DOI:10.1039/C6GC02271E |
| [17] |
X. Ji, Z.W Dongdong, et al., Asian J. Org. Chem. 7 (2018) 711-714. DOI:10.1002/ajoc.201800036 |
| [18] |
Z. Wang, X. Ji, J. Zhao, et al., Green Chem. 21 (2019) 5512-5516. DOI:10.1039/C9GC03008E |
| [19] |
L. Tang, K. Du, B. Yu, et al., Chin. Chem. Lett. 31 (2020) 2991-2992. DOI:10.1016/j.cclet.2020.03.030 |
| [20] |
L. Yao, D. Zhu, L. Wang, et al., Chin. Chem. Lett. 32 (2021) 4033-4037. DOI:10.1016/j.cclet.2021.06.005 |
| [21] |
H. Sterckx, B. Morel, B.U.W. Maes, Angew. Chem. Int. Ed. 58 (2019) 7946-7970. DOI:10.1002/anie.201804946 |
| [22] |
D. Wang, A.B. Weinstein, P.B. White, et al., Chem. Rev. 118 (2018) 2636-2679. DOI:10.1021/acs.chemrev.7b00334 |
| [23] |
H. Huang, X. Ji, X. Zhou, et al., Chin. J. Org. Chem. 41 (2021) 4704-4711. DOI:10.6023/cjoc202111037 |
| [24] |
J.M. Narayanam, C.R. Stephenson, Chem. Soc. Rev. 40 (2011) 102-113. DOI:10.1039/B913880N |
| [25] |
J. Xuan, W.J. Xiao, Angew. Chem. Int. Ed. 51 (2012) 6828-6838. DOI:10.1002/anie.201200223 |
| [26] |
C.K. Prier, D.A. Rankic, D.W. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [27] |
N. Corrigan, S. Shanmugam, J. Xu, et al., Chem. Soc. Rev. 45 (2016) 6165-6212. DOI:10.1039/C6CS00185H |
| [28] |
W.B. He, L.Q. Gao, X.J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1895-1898. DOI:10.1016/j.cclet.2020.02.011 |
| [29] |
J. Xuan, B. Cai, Chin. J. Org. Chem. 41 (2021) 4565-4574. DOI:10.6023/cjoc202109040 |
| [30] |
B. Yu, Q. Lü, X. Chen, et al., Chin. J. Org. Chem. 41 (2021) 4575-4587. DOI:10.6023/cjoc202109008 |
| [31] |
S. Mohle, M. Zirbes, E. Rodrigo, et al., Angew. Chem. Int. Ed. 57 (2018) 6018-6041. DOI:10.1002/anie.201712732 |
| [32] |
S. Tang, Y. Liu, A. Lei, Chem 4 (2018) 27-45. DOI:10.1016/j.chempr.2017.10.001 |
| [33] |
S.R. Waldvogel, S. Lips, M. Selt, et al., Chem. Rev. 118 (2018) 6706-6765. DOI:10.1021/acs.chemrev.8b00233 |
| [34] |
J. Jiang, Z. Wang, W.M. He, Chin. Chem. Lett. 32 (2021) 1591-1592. DOI:10.1016/j.cclet.2021.02.067 |
| [35] |
J. Li, S. Zhang, K. Xu, Chin. Chem. Lett. 32 (2021) 2729-2735. DOI:10.1016/j.cclet.2021.03.027 |
| [36] |
W. He, J. Chen, Y. Lu, et al., Chin. J. Org. Chem. 41 (2021) 4766-4772. DOI:10.6023/cjoc202109044 |
| [37] |
B. Zhang, J. Meng, K. Sun, et al., Chin. J. Org. Chem. 41 (2021) 4588-4609. DOI:10.6023/cjoc202109046 |
| [38] |
D.S. Bolotin, N.A. Bokach, M.Y. Demakova, et al., Chem. Rev. 117 (2017) 13039-13122. DOI:10.1021/acs.chemrev.7b00264 |
| [39] |
A.A. Tabolin, S.L. Ioffe, Chem. Rev. 114 (2014) 5426-5476. DOI:10.1021/cr400196x |
| [40] |
M. Kitamura, K. Narasaka, Chem. Rec. 2 (2002) 268-277. DOI:10.1002/tcr.10030 |
| [41] |
K. Narasaka, M. Kitamura, Eur. J. Org. Chem. 2005 (2005) 4505-4519. DOI:10.1002/ejoc.200500389 |
| [42] |
W. Li, T.B. Towne, G. Liang, et al., Org. Process Res. Dev. 18 (2014) 1696-1701. DOI:10.1021/op500247h |
| [43] |
W. Xiao, J. Wu, Chin. Chem. Lett. 31 (2020) 3083-3094. DOI:10.1016/j.cclet.2020.07.035 |
| [44] |
H. Huang, X. Ji, W. Wu, et al., Chem. Soc. Rev. 44 (2015) 1155-1171. DOI:10.1039/C4CS00288A |
| [45] |
S. Choi, S. Ha, C.M. Park, Chem. Commun. 53 (2017) 6054-6064. DOI:10.1039/C7CC02650A |
| [46] |
J. Li, Y. Hu, D. Zhang, et al., Adv. Synth. Catal. 359 (2017) 710-771. DOI:10.1002/adsc.201600807 |
| [47] |
H. Huang, J. Cai, G.J. Deng, Org. Biomol. Chem. 14 (2016) 1519-1530. DOI:10.1039/C5OB02417J |
| [48] |
C. Chen, J. Zhao, X. Shi, et al., Org. Chem. Front. 7 (2020) 1948-1969. DOI:10.1039/D0QO00397B |
| [49] |
D. Ravelli, D. Dondi, M. Fagnoni, et al., Chem. Soc. Rev. 38 (2009) 1999-2011. DOI:10.1039/b714786b |
| [50] |
T.P. Yoon, M.A. Ischay, J. Du, Nat. Chem. 2 (2010) 527-532. DOI:10.1038/nchem.687 |
| [51] |
J. Jiang, F. Xiao, W.M. He, et al., Chin. Chem. Lett. 32 (2021) 1637-1644. DOI:10.1016/j.cclet.2021.02.057 |
| [52] |
A.A. Festa, L.G. Voskressensky, E.V. Van der Eycken, Chem. Soc. Rev. 48 (2019) 4401-4423. DOI:10.1039/C8CS00790J |
| [53] |
S. Liu, F. Zhao, X. Chen, et al., Adv. Synth. Catal. 362 (2020) 3795-3823. DOI:10.1002/adsc.202000285 |
| [54] |
J.S.S. Neto, G. Zeni, Org. Chem. Front. 7 (2020) 155-210. DOI:10.1039/C9QO01315F |
| [55] |
H. Huang, J. Cai, X. Ji, et al., Angew. Chem. Int. Ed. 55 (2016) 307-311. DOI:10.1002/anie.201508076 |
| [56] |
H. Huang, F. Li, Z. Xu, et al., Adv. Synth. Catal. 359 (2017) 3102-3107. DOI:10.1002/adsc.201700730 |
| [57] |
X. Tang, L. Huang, J. Yang, et al., Chem. Commun. 50 (2014) 14793-14796. DOI:10.1039/C4CC06747A |
| [58] |
Q. Wu, Y. Zhang, S. Cui, Org. Lett. 16 (2014) 1350-1353. DOI:10.1021/ol500094w |
| [59] |
H. Huang, X. Ji, X. Tang, et al., Org. Lett. 15 (2013) 6254-6257. DOI:10.1021/ol403105p |
| [60] |
Z. Zhu, X. Tang, J. Li, et al., Org. Lett. 19 (2017) 1370-1373. DOI:10.1021/acs.orglett.7b00203 |
| [61] |
E.J. Miller, E. Jecs, V.M. Truax, et al., J. Med. Chem. 61 (2018) 946-979. DOI:10.1021/acs.jmedchem.7b01420 |
| [62] |
Y. Fang, H. Zhou, Q. Gu, et al., Eur. J. Med. Chem. 167 (2019) 133-145. DOI:10.1016/j.ejmech.2019.02.008 |
| [63] |
Z. Qu, F. Zhang, G.J. Deng, et al., Org. Lett. 21 (2019) 8239-8243. DOI:10.1021/acs.orglett.9b02978 |
| [64] |
A. Ibrar, I. Khan, N. Abbas, et al., RSC Adv. 6 (2016) 93016-93047. DOI:10.1039/C6RA19324B |
| [65] |
F. Xiao, S. Yuan, H. Huang, et al., Org. Lett. 21 (2019) 8533-8536. DOI:10.1021/acs.orglett.9b02934 |
| [66] |
X. Tang, L. Huang, C. Qi, et al., Chem. Commun.. |
| [67] |
Y.Y. Yu, A.R. Ranade, G.I. Georg, Adv. Synth. Catal. 356 (2014) 3510. DOI:10.1002/adsc.201400417 |
| [68] |
G.C. Senadi, T.Y. Lu, G.K. Dhandabani, et al., Org. Lett. 19 (2017) 1172-1175. DOI:10.1021/acs.orglett.7b00208 |
| [69] |
H.B. Yang, N. Selander, Chem. Eur. J. 23 (2017) 1779-1783. DOI:10.1002/chem.201605636 |
| [70] |
B. Zhao, H.W. Liang, J. Yang, et al., ACS Catal. 7 (2017) 5612-5617. DOI:10.1021/acscatal.7b01876 |
| [71] |
Y. Xie, Y. Li, X. Chen, et al., Org. Chem. Front. 5 (2018) 1698-1701. DOI:10.1039/C8QO00204E |
| [72] |
Y. Cai, A. Jalan, A.R. Kubosumi, et al., Org. Lett. 17 (2015) 488-491. DOI:10.1021/ol5035047 |
| [73] |
Z. Xu, N. Xian, H. Chen, et al., Chin. J. Chem. 39 (2021) 1175-1180. DOI:10.1002/cjoc.202000660 |
| [74] |
Z.H. Ren, Z.Y. Zhang, B.Q. Yang, et al., Org. Lett. 13 (2011) 5394-5397. DOI:10.1021/ol202290y |
| [75] |
H. Jiang, J. Yang, X. Tang, et al., J. Org. Chem. 80 (2015) 8763-8771. DOI:10.1021/acs.joc.5b01621 |
| [76] |
M.N. Zhao, L. Yu, N.F. Mo, et al., Org. Chem. Front. 4 (2017) 597-602. DOI:10.1039/C6QO00809G |
| [77] |
A. Ramaraju, N.K. Chouhan, O. Ravi, et al., Eur. J. Org. Chem. 2018 (2018) 2963-2971. DOI:10.1002/ejoc.201800501 |
| [78] |
J.L. Zhan, M.W. Wu, D. Wei, et al., ACS Catal. 9 (2019) 4179-4188. DOI:10.1021/acscatal.9b00832 |
| [79] |
Y. Wei, N. Yoshikai, J. Am. Chem. Soc. 135 (2013) 3756-3759. DOI:10.1021/ja312346s |
| [80] |
W.W. Tan, Y.J. Ong, N. Yoshikai, Angew. Chem. Int. Ed. 56 (2017) 8240-8244. DOI:10.1002/anie.201704378 |
| [81] |
D. Bai, X. Wang, G. Zheng, et al., Angew. Chem. 130 (2018) 6743-6747. DOI:10.1002/ange.201802311 |
| [82] |
H. Huang, J. Cai, L. Tang, et al., J. Org. Chem. 81 (2016) 1499-1505. DOI:10.1021/acs.joc.5b02624 |
| [83] |
H. Huang, J. Cai, H. Xie, et al., Org. Lett. 19 (2017) 3743-3746. DOI:10.1021/acs.orglett.7b01564 |
| [84] |
Y. Xia, J. Cai, H. Huang, et al., Org. Biomol. Chem. 16 (2018) 124-129. DOI:10.1039/C7OB02471A |
| [85] |
Z. Xu, H. Chen, G.J. Deng, et al., Org. Lett. 23 (2021) 936-942. DOI:10.1021/acs.orglett.0c04138 |
| [86] |
X. Tang, J. Yang, Z. Zhu, et al., J. Org. Chem. 81 (2016) 11461-11466. DOI:10.1021/acs.joc.6b02124 |
| [87] |
X. Tang, Z. Zhu, C. Qi, et al., Org. Lett. 18 (2016) 180-183. DOI:10.1021/acs.orglett.5b03188 |
| [88] |
Z. Zhu, X. Tang, J. Cen, et al., Chem. Commun. 54 (2018) 3767-3770. DOI:10.1039/C8CC00445E |
| [89] |
D.A. Boyd, Angew. Chem. Int. Ed. 55 (2016) 15486-15502. DOI:10.1002/anie.201604615 |
| [90] |
K. Takimiya, I. Osaka, T. Mori, et al., Acc. Chem. Res. 47 (2014) 1493-1502. DOI:10.1021/ar400282g |
| [91] |
H. Huang, G.J. Deng, S. Liu, Synlett 32 (2020) 142-158. |
| [92] |
H. Huang, Z. Xu, X. Ji, et al., Org. Lett. 20 (2018) 4917-4920. DOI:10.1021/acs.orglett.8b02049 |
| [93] |
H. Huang, Q. Wang, Z. Xu, et al., Adv. Synth. Catal. 361 (2019) 591-596. DOI:10.1002/adsc.201801324 |
| [94] |
H. Huang, Z. Qu, X. Ji, et al., Org. Chem. Front. 6 (2019) 1146-1150. DOI:10.1039/C8QO01365A |
| [95] |
Y. Chen, S. Lv, R. Lai, et al., Chin. Chem. Lett. 32 (2021) 2555-2558. DOI:10.1016/j.cclet.2021.02.052 |
| [96] |
Z. Xu, G.J. Deng, F. Zhang, et al., Org. Lett. 21 (2019) 8630-8634. DOI:10.1021/acs.orglett.9b03241 |
| [97] |
K. Deng, H. Huang, G.J. Deng, Org. Biomol. Chem. 19 (2021) 6380-6391. DOI:10.1039/D1OB00908G |
| [98] |
G. Deng, M. Yang, R. Li, et al., Chin. J. Org. Chem. 42 (2022) 129-146. DOI:10.6023/cjoc202107032 |
| [99] |
Z. Xu, H. Huang, H. Chen, et al., Org. Chem. Front. 6 (2019) 3060-3064. DOI:10.1039/C9QO00592G |
| [100] |
H. Meng, Z. Xu, Z. Qu, et al., Org. Lett. 22 (2020) 6117-6121. DOI:10.1021/acs.orglett.0c02180 |