 2022, Vol. 33
2022, Vol. 33 


b Hubei Key Laboratory of Low Dimensional Optoelectronic Materials and Devices, Hubei University of Arts and Science, Xiangyang 441053, China;
c Institute of Mathematics, Free University of Berlin, Berlin D-14195, Germany
N2 fixation for NH3 synthesis is of great significance since NH3 is one of the main feedstocks for chemical and agricultural industries and also a promising carbon-free energy carrier with high energy density for the future economy [1-5]. Compared with the conventional energy- and resource-intensive Haber-Bosch process, electrocatalytic N2 reduction reaction (NRR), which hydrogenates N2 as well as protons and electrons at ambient conditions, is regarded as a more cost-effective, sustainable, and environmentally benign method for NH3 synthesis [6-8]. However, as a nonpolar homonuclear diatomic molecule, N2 is fairly inert due to the extremely thermodynamically stable covalent triple bond (941 kJ/mol) and thus is difficult to break in the NRR process, resulting in low N2-to-NH3 conversion efficiency [9-12]. Consequently, the rational design of efficient NRR electrocatalysts for NH3 synthesis is highly desirable but remains great challenge.
Today, single-atom catalysts (SACs) comprising isolated atoms dispersed on substrates are of particular interest in the field of electrocatalysis due to their exceptional catalytic performances and maximized atomic utilization [13-15]. In particular, many SACs of transition-metal (TM) atoms anchored with two-dimensional (2D) materials, including graphene [16, 17], h-BN [18, 19], borophene [20, 21], phosphorene [22-24], graphyne [25, 26], porous carbon-nitride nanosheets [27-30], nitrogen-doped carbon [31, 32] and so on [33-35], have been theoretically predicted as striking NRR electrocatalysts with ultralow limiting potential. However, as multiple reaction intermediates are typically involved in the NRR process, experimental studies reveal that the single active site is still rather hard to break the scaling relations and enhance both the yield rate of NH3 and the Faradaic efficiency (FE) simultaneously. To address this issue, much effort has been devoted to designing double-atom catalysts (DACs), which possess more flexible active sites to tune the adsorption behavior of NRR intermediates [36-40]. For instance, Li et al. [41] synthesized atomically dispersed Fe-Mo dimers anchored on N-doped graphene (FeMo@NG) and observed that it exhibited better NRR catalytic activity than its SAC counterparts (Fe@NG and Mo@NG) due to the combination of geometric, ligand and synergistic effects. Han et al. [42] reported that the as-fabricated Pd-Cu diatom catalyst had higher FE and NH3 yield rate than the individual single-atom (Pd or Cu) catalyst. In addition, theoretical screening and designing based on first-principles calculations found that Mo-, Fe-, Co-, Mn- and Ru-based DACs can be served as excellent catalysts for electrochemical N2 reduction [43-55].
Recently, a novel porous graphitic carbon nitride (g-C6N6) is successfully synthesized via the solvothermal reaction of C3N3Cl3 with Na [56]. Interestingly, g-C6N6 possesses a high specific surface area, excellent thermal and kinetic stability, and outstanding electric conductivity, making it a remarkable substrate to anchor TM atoms for electrocatalytic reactions [57-59]. Inspired by these exciting progressive achievements, in the present work, a series of single and double TM atoms (TM = Fe, Co, Ni and Mo) anchored on g-C6N6 are designed and explored as NRR electrocatalysts. Among them, Mo2@g-C6N6 is screened out as the most promising candidate for electrochemical NRR due to its high stability, activity, and selectivity. Our work indicates that DACs have great chance to replace SACs as the emerging star of atomic catalysts.
All the spin-polarized first-principles calculations are performed using the density functional theory (DFT), as implemented in Vienna ab initio Simulation Package (VASP) [60]. The interactions between electrons and ion-cores are described by the projector augmented wave (PAW) method [61] and the exchange-correlation interactions are treated by the generalized gradient approximation (GGA) in the form proposed by Perdew, Burke, and Ernzerhof (PBE) [62]. Additionally, Grimme's semiempirical DFT-D3 method is employed to address the weak van der Waals interactions between the adsorbates and substrate [63]. The cut-off energy is set to 500 eV for the plane-wave basis in all of our calculations. Γ-centered k-point meshes of 2 × 2 × 1 and 4 × 4 × 1 based on Monkhorst-Pack scheme [64] are employed for the geometric and electronic structures calculations, respectively. A vacuum space of 20.0 Å along the z-axis is adopted to ensure no appreciable interaction between the image layers under periodic boundary condition. The self-consistent convergence criterion for the total energy and Hellmann-Feynman force are smaller than 10−5 eV and 0.02 eV/Å, respectively. More computational details for the NRR are given in the Supporting information.
The optimized geometrical structure of pristine g-C6N6 monolayer is depicted in Fig. 1a, which contains six carbon atoms and six nitrogen atoms in the hexagonal unit cell with space group P6/MMM and point group D6H-1. The calculated lattice constants are found to be a = b = 7.12 Å with the C—C and C—N bond distances of 1.51 and 1.34 Å, respectively, in good agreement with previous reports [65-67]. In this study, a 2 × 2 × 1 g-C6N6 supercell is employed as an ideal substrate for anchoring the selected four types of common TM atoms (TM = Fe, Co, Ni and Mo). To find the most favorable anchoring sites, we examine two possible configurations of the single and double TM atoms on the g-C6N6 monolayer (labeled as TM@g-C6N6 and TM2@g-C6N6, respectively), in which TM atoms bond with two N atoms or three N atoms of g-C6N6 substrate. As shown in Fig. 1b, we find that the single TM atom bonds strongly with adjacent two N atoms at the cavity edge after full optimization except for the Mo atom, while all the double TM atoms are located within the six-remember nitrogen hole and bond with three N atoms. The distances of TM-TM bonds are 2.13 Å for Fe, 2.18 Å for Co, 2.34 Å for Ni, and 1.78 Å for Mo. The planar structure of the g-C6N6 monolayer can be maintained well before and after anchoring these TM atoms. Compared to pristine g-C6N6 with a band gap of 1.83 eV (Fig. S1 in Supporting information), these TMn@g-C6N6 monolayers exhibit metallic features mainly originated from the hybridized states between TM-d and the adjacent N-p orbitals (Fig. S2 in Supporting information), which will be propitious to the electrocatalytic reactions.

|
Download:
|
| Fig. 1. (a) The atomic structure of pristine g-C6N6 monolayer. The rhombus denotes the unit cell, and the brown and light-blue spheres stand for C and N atoms, respectively. (b) The optimized most stable configuration and (c) calculated binding energies of TM@g-C6N6 and TM2@g-C6N6. | |
{kind=link}
To estimate the structural stability of TMn@g-C6N6, we calculate the binding energies (Eb), as shown in Fig. 1c. Overall, all double TM anchored on g-C6N6 show larger binding energies compared to the corresponding single TM anchored cases, suggesting that the formed metal-metal bonding can offer positive effects on stabilization of these TM dimers. Moreover, the Eb of TMn@g-C6N6 are more negative than their respective cohesive energies of bulk metal, signifying strong interactions between the TM atoms and g-C6N6 monolayer, and superior thermodynamic stability. In addition, we further check the temperature-dependent stability of TMn@g-C6N6 by ab initio molecular dynamics (AIMD) simulations as depicted in Fig. S3 (Supporting information). We observe that these TMn@g-C6N6 systems display narrow energy fluctuations and slight structural changes during the simulated time, verifying the excellent thermal stability at room temperature. Thus, it is believed that the as-designed TMn@g-C6N6 could serve as potential electrocatalyst with high stability.
Following the screening requirements for NRR catalyst [68, 69], we turn to examine the Gibbs free energy changes (△G) of the N2 adsorption and first hydrogenation process of NRR to produce *N2H on TMn@g-C6N6 for identifying the most qualified candidate. The computed △G for the two processes are presented in Figs. 2a and b, respectively. Our results show that all the TM@g-C6N6 systems are incapable of capturing N2 molecule through the side-on configuration because of their positive △G*N, whereas N2 molecule can effectively chemisorb on TM@g-C6N6 by means of the end-on configuration. However, the first protonation of adsorbed N2 (*N2 + H+ + e→ *N2H) require much high energy inputs, such as 1.34 eV for Fe@g-C6N6, 1.33 eV for Co@g-C6N6, 1.25 eV for Ni@g-C6N6, and 0.51 eV for Mo@g-C6N6, demonstrating that they are inefficient electrocatalysts for N2 reduction. In this regard, the TM@g-C6N6 systems are eliminated as eligible NRR catalysts.

|
Download:
|
| Fig. 2. Calculated Gibbs free energies (△G) for (a) N2 adsorption and (b) *N2H formation on TM@g-C6N6 and TM2@g-C6N6. | |
{kind=link}
With respect to TM@g-C6N6, we find that except for Ni2@g-C6N6, N2 molecule can be well captured by TM2@g-C6N6 regardless of end-on or side-on configuration, as indicated by the calculated △G*N ranging from −0.46 eV to −0.81 eV. Further, the △G values of *N2H formation on the Fe2@g-C6N6 and Co2@g-C6N6 are predicted to be at least 0.48 and 0.72 eV, respectively, marking them as inefficient electrocatalysts for N2 reduction. Notably, the first hydrogenation process of NRR to produce *N2H on Mo2@g-C6N6 demands only 0.11 eV energy input for the side-on configuration, while it even becomes a spontaneous reaction for the end-on configuration. Based on these screening results, only the Mo2@g-C6N6 is of interest in virtue of satisfying all the requirements, and thus will be examined in the following discussion.
Figs. 3a and b depict the optimized structures of N2 adsorbed Mo2@g-C6N6 via end-on and side-on configurations, respectively. Compared with the isolated N2 molecule (1.12 Å), the N—N bond length is significantly stretched by 0.04 Å for end-on adsorption and by 0.09 Å for side-on adsorption, signifying the effective activation of the inert triple bond of N2. Bader charge analysis shows that there are about 0.49 and 0.70 electrons transferred from Mo2@g-C6N6 to the adsorbed N2 in end-on and side-on patterns, respectively, which attributes to the different electronegativities of N (3.04) and Mo (1.86). Moreover, as presented in Figs. 3c and d, we can observe the obvious charge accumulation and depletion around the anchored Mo dimer and N2 for both adsorption configurations. Interestingly, such a charge distribution ideally accords with the "acceptance-donation" process as proposed by Ling and co-workers [70], that is, the Mo dimer donates electrons into the antibonding orbitals of N2 and simultaneously accepts lone-pair electrons from it. From the computed projected density of states given in Figs. 3e and f, we find that there are significant electronic coupling between the N-2p orbital of the adsorbed N2 molecule and Mo-4d orbital, thus give rise to the anti-bonding states located above the Fermi level and bonding states lied below the Fermi level, further supporting the "acceptance-donation" picture. Moreover, after N2 adsorption with side-on configuration, the magnetic moments of the Mo dimer and N2 molecule are nearly unchanged and remain 0 µB. However, for N2 adsorption with end-on configuration, we observe that their corresponding magnetic moments increase to 0.97 µB and 0.24 µB, respectively. As shown in Fig. S4 (Supporting information), the increased magnetic moments attribute to the asymmetrical distributions of Mo-4d and N-2p orbitals, caused by the couplings of Mo 4dz-N 2py, Mo 4dxz-N 2pz and Mo 4dznullN 2pz. These above analyses demonstrate the successful activation of N2 on Mo2@g-C6N6, which would further facilitate the subsequent reaction of converting N2 to NH3 molecules.

|
Download:
|
| Fig. 3. (a) End-on and (b) side-on adsorption configurations for N2 on Mo2@g-C6N6 monolayer. (c, d) The corresponding charge density difference, where the isosurface value is set to be 0.005 e/Å3 and the yellow and cyan show the positive and negative charges, respectively. The computed projected density of states of Mo2@g-C6N6 with the N2 adsorption through (e) end-on and (f) side-on configurations. | |
{kind=link}
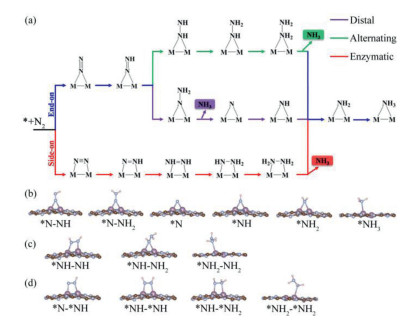
As illustrated in Fig. 4a, three possible reaction mechanisms, named distal, alternating, and enzymatic, are considered for the transformation from N2 to NH3. Both distal and alternating mechanisms start from the adsorbed N2 with end-on configuration, while the enzymatic mechanism is initiated with the side-on adsorbed N2. Whereafter, each mechanism involves six consecutive protonation plus reduction steps, accompanied by the production of two NH3 molecules. The optimized geometries of all reaction intermediates along these three pathways on Mo2@g-C6N6 monolayer are given in Figs. 4b-d, and the predicted Gibbs free energy profiles are illustrated in Figs. 5a-c.

|
Download:
|
| Fig. 4. (a) Schematic depiction of three possible reaction mechanisms for N2 electroreduction to NH3 on the Mo2@g-C6N6 monolayer. Optimized geometries of all reaction intermediates along the (b) distal, (c) alternating, and (d) enzymatic pathways. | |
{kind=link}

|
Download:
|
| Fig. 5. Gibbs free energy profiles of N2 reduction on Mo2@g-C6N6 monolayer via the (a) distal, (b) alternating and (c) enzymatic mechanisms at zero and applied potentials. (d) Charge variation of three moieties for the NRR along the preferred distal pathway. | |
{kind=link}
As NRR proceeds along the distal or alternating pathway, the first protonation plus reduction step is similar, and found to be exothermic by 0.02 eV with the further elongation of N—N bond (from 1.16 Å to 1.25 Å). It is noteworthy that this first hydrogenation is normally non-spontaneous for most studied electrocatalysts since the free energy change is thermodynamically uphill. For the following elementary steps through distal pathway, the *N—NH can be spontaneously hydrogenated to *NH2 by reacting with the proton-electron pair four times. Specifically, as shown in Fig. 5a, the Gibbs reaction free energies of these four hydrogenation steps are −0.10, −0.22, −0.80 and −0.81 eV, respectively. Finally, the process of *NH2 + H+ + e− → *NH3 is slightly uphill by 0.06 eV in the Gibbs free energy profile. Overall, the potential-limiting step (PDS) of the distal mechanism is the sixth protonation plus reduction step owing to its maximum △G of 0.06 eV. For the case of the alternating mechanism, as given in Fig. 5b, the Gibbs free energy changes along the subsequent protonation plus reduction steps are calculated respectively to be −0.02, 0.07, −0.34, 0.22, −1.88 and 0.06 eV. Thus, the hydrogenation of *NH—NH2 into *NH2nullNH2 is the PDS with a maximal energy demand of 0.22 eV. Regarding of the enzymatic mechanism, the first protonation plus reduction step is slightly uphill energetically, with a △G value of 0.11 eV as demonstrated in Fig. 5c. After that, the subsequent elementary reaction steps are all exothermic, except for the formation of *NH-*NH2 (*NH-*NH + H+ + e− → *NH-*NH2) and second NH3 (*NH2 + H+ + e− → *NH3). Among the hydrogenation steps of the enzymatic mechanism, the step converting *N-*NH to *NH-*NH2 has the largest free energy change (0.28 eV), which can be identified as the PDS. Of note, the desorption of the second NH3 from Mo2@g-C6N6 monolayer is not a problematic obstacle. As demonstrated in previous studies [55, 66], the *NH3 species can be released in the form of NH4+ under acidic electrochemical conditions. Moreover, the free energy change for the release of the final product NH3 is only 0.82 eV, which can be easily overcame in consideration of the released energy (about 2.60 eV, Fig. 5) in the proceeding hydrogenation steps. These ensure the rapid removal of the produced NH3 under ambient conditions for recovering the catalysts.
In electrocatalysis, the limiting potential (UL), defined as the lowest applied electrode potential to overcome the maximum positive △G on the considered pathway, is the most commonly used descriptor for chemical reactivity, which can be calculated by UL = −△Gmax/e [71, 72]. A less negative UL indicates a faster electrochemical process at a given potential. As discussed above, the limiting potential with respect to the standard hydrogen electrode are calculated to be −0.06 V for distal, −0.22 V for alternating, and −0.28 V for enzymatic. Consequently, we predict that the NRR on Mo2@g-C6N6 will prefer to proceed via the distal mechanism in virtue of its lowest negative value. Remarkably, such a limiting potential is substantially less negative than the equilibrium potential of overall NRR (−0.16 V) as well as other recently reported DACs for NH3 synthesis, implying that the applied potential even as low as the equilibrium potential can adequately drive the NRR. Therefore, Mo2@g-C6N6 could be expected as a promising candidate electrocatalyst for N2 reduction to NH3.
It should be pointed out that conventional DFT method often fails to describe the systems with strongly correlated d-electrons. In order to further evaluate the reliability of the above results, we employ the DFT+U method to re-examine Gibbs free energy profile of N2 reduction on Mo2@g-C6N6 monolayer through the preferred distal pathway. As shown in Fig. S5 (Supporting information), we can see that the Hubbard U value has little influence on the free energy diagram, and both of the limiting potential and PDS with the Hubbard U correction are in line with the PBE results, suggesting that the PBE calculated results are acceptable in this work.
To gain insight into the superior catalytic activity of Mo2@g-C6N6, we further employ Bader charge analysis to investigate the charge variation of various intermediates along the favorable distal pathway. Herein, we divide each intermediate into three groups, including the g-C6N6, anchored Mo dimer, and adsorbed NxHy species. As depicted in Fig. 5d, the N2 molecule after chemisorption on the Mo dimer accumulates about 0.49 electrons, which is considered to be propitious for the hydrogeneration of *N2 into *N2H. In the following NRR steps, it is observed that there are obvious charge fluctuations in both of the g-C6N6 and NxHy groups, while the charge value of Mo dimer remains almost unchanged. That is to say, the charge variation of the NxHy species mainly associates with that of the g-C6N6. The Mo dimer serves as not only the active site but also electron transmitter between the g-C6N6 and NxHy species during the entire NRR process.
As one of the prerequisites of an excellent NRR electrocatalyst with high FE, it is crucial to suppress the hydrogen evolution reaction (HER), which is the major competing reaction during the NRR. Therefore, we compute the adsorption free energies of *H (△G*H) at the Mo dimer site of Mo2@g-C6N6 monolayer. The △G*H is found to be 0.46 eV, which is more positive than the △G*N (−0.71 eV), suggesting that the active site would be primarily covered by *N2. Moreover, the free energy barrier for HER (0.46 eV) is considerable larger than the PDS barrier for NRR (0.06 eV). In this respect, the FE of Mo2@g-C6N6 is estimated to be approximately 100% at room temperature according to the Boltzmann distribution, indicating the high selectivity for NRR.
In conclusion, by performing comprehensive DFT computations, we explore the potential of single and double transition metal atoms (TM = Fe, Co, Ni and Mo) anchored g-C6N6 monolayers for electrochemical N2 reduction to NH3. Among these systems, Mo2@g-C6N6 monolayer is identified as the most promising catalyst candidate toward NRR with the help of prescreening criteria. Our results demonstrate that N2 molecule is effectively captured and activated by the Mo dimer anchored on g-C6N6 through the "acceptance-donation" process. The subsequent N2 reduction reaction on Mo2@g-C6N6 monolayer proceed dominantly via the distal mechanism with an extremely low limiting potential of −0.06 V. Importantly, the competitive HER can be well inhibited on the Mo2@ g-C6N6 monolayer due to a much larger △G*H, ensuring substantial selectivity (~100%) toward NH3 synthesis. Hence, the Mo2@g-C6N6 monolayer proposed in this work has great potential applications for NRR, highlighting the importance of DACs for NRR.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Science & Technology Development Fund of Tianjin Education Commission for Higher Education (No. 2020KJ008), by the Natural Science Foundation of Tianjin (No. 18JCQNJC76000), and by the College Students' Innovation and Entrepreneurship Training Program of Tianjin (No. 202110065112), Science and Technology Research Project of Hubei Provincial Department of Education (No. D20212603), Hubei University of Arts and Science (Nos. 2020kypytd002, XK2021024) and China Scholarship Council.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.040.
| [1] |
O. Elishav, B. Mosevitzky Lis, E.M. Miller, et al., Chem. Rev. 120 (2020) 5352-5436. DOI:10.1021/acs.chemrev.9b00538 |
| [2] |
L. Zhao, Y. Li, G. Zhou, et al., Chin. Chem. Lett. 32 (2021) 900-905. DOI:10.1016/j.cclet.2020.07.016 |
| [3] |
Y. Yang, Y. Tang, H. Jiang, et al., Chin. Chem. Lett. 30 (2019) 2089-2109. DOI:10.1016/j.cclet.2019.10.041 |
| [4] |
J. Liu, J. Ma, Z. Zhang, et al., J. Phys. Mater. 4 (2021) 022004. DOI:10.1088/2515-7639/abd596 |
| [5] |
J. Wang, J. Teng, L. Pu, et al., Int. J. Quantum Chem. 119 (2019) e25930. DOI:10.1002/qua.25930 |
| [6] |
J. Sun, W. Kong, Z. Jin, et al., Chin. Chem. Lett. 31 (2020) 953-960. DOI:10.1016/j.cclet.2020.01.035 |
| [7] |
Y. Wang, Q. Li, W. Shi, et al., Chin. Chem. Lett. 31 (2020) 1768-1772. DOI:10.1016/j.cclet.2020.01.010 |
| [8] |
S. Li, M. Shi, J. Yu, et al., Chin. Chem. Lett. 32 (2021) 1977-1982. DOI:10.1016/j.cclet.2020.09.056 |
| [9] |
B.H.R. Suryanto, H.L. Du, D. Wang, et al., Nat. Catal. 2 (2019) 290-296. DOI:10.1038/s41929-019-0252-4 |
| [10] |
G. Qing, R. Ghazfar, S.T. Jackowski, et al., Chem. Rev. 120 (2020) 5437-5516. DOI:10.1021/acs.chemrev.9b00659 |
| [11] |
Y. Ren, C. Yu, X. Tan, et al., Energy Environ. Sci. 14 (2021) 1176-1193. DOI:10.1039/d0ee03596c |
| [12] |
J. Wang, S. Li, F. Yun, et al., Int. J. Quantum Chem. 120 (2020) e26230. |
| [13] |
T. Sun, S. Mitchell, J. Li, et al., Adv. Mater. 33 (2021) 2003075. DOI:10.1002/adma.202003075 |
| [14] |
S. Yuan, B. Xu, S. Li, et al., Chin. Chem. Lett. 33 (2022) 399-403. DOI:10.1016/j.cclet.2021.06.077 |
| [15] |
J. Fu, K. Liu, K. Jiang, et al., Adv. Sci. 6 (2019) 1900796. DOI:10.1002/advs.201900796 |
| [16] |
P. Liu, C. Fu, Y. Li, et al., Phys. Chem. Chem. Phys. 22 (2020) 9322-9329. DOI:10.1039/c9cp06112f |
| [17] |
Z. Wei, Y. Feng, J. Ma, J. Energy Chem. 48 (2020) 322-327. DOI:10.1016/j.jechem.2020.02.014 |
| [18] |
J. Zhao, Z. Chen, J. Am. Chem. Soc. 139 (2017) 12480-12487. DOI:10.1021/jacs.7b05213 |
| [19] |
Z. Ma, Z. Cui, C. Xiao, et al., Nanoscale 12 (2020) 1541-1550. DOI:10.1039/c9nr08969a |
| [20] |
L. Xu, L.M. Yang, E. Ganz, ACS Appl. Mater. Interfaces 13 (2021) 14091-14101. DOI:10.1021/acsami.0c20553 |
| [21] |
X. Kang, Z. Chu, X. Duan, Appl. Surf. Sci. 560 (2021) 149667. DOI:10.1016/j.apsusc.2021.149667 |
| [22] |
Z.M. Zhang, X. Yao, X.Y. Lang, et al., Appl. Surf. Sci. 536 (2021) 147706. DOI:10.1016/j.apsusc.2020.147706 |
| [23] |
J.D. Liu, Z.X. Wei, Y.H. Dou, et al., Rare Met. 39 (2020) 874-880. DOI:10.1007/s12598-020-01451-z |
| [24] |
K. Liu, J. Fu, L. Zhu, et al., Nanoscale 12 (2020) 4903-4908. DOI:10.1039/c9nr09117c |
| [25] |
T. He, S.K. Matta, A. Du, Phys. Chem. Chem. Phys. 21 (2019) 1546-1551. DOI:10.1039/c8cp06978f |
| [26] |
W. Song, L. Fu, C. He, et al., Adv. Theory Simul. 4 (2021) 2100044. DOI:10.1002/adts.202100044 |
| [27] |
S. Wang, W. Wei, X. Lv, et al., J. Mater. Chem. A 8 (2020) 1378-1385. DOI:10.1039/c9ta10935h |
| [28] |
H. Niu, X. Wang, C. Shao, et al., ACS Sustain. Chem. Eng. 8 (2020) 13749-13758. DOI:10.1021/acssuschemeng.0c04401 |
| [29] |
Z. Xue, X. Zhang, J. Qin, et al., J. Energy Chem. 57 (2021) 443-450. DOI:10.1016/j.jechem.2020.09.002 |
| [30] |
J. Wang, M. Shi, G. Yi, et al., Mol. Catal. 511 (2021) 111726. DOI:10.1016/j.mcat.2021.111726 |
| [31] |
D. Jiao, Y. Liu, Q. Cai, et al., J. Mater. Chem. A 9 (2021) 1240-1251. DOI:10.1039/d0ta09496j |
| [32] |
W. Zhao, L. Chen, W. Zhang, et al., J. Mater. Chem. A 9 (2021) 6547-6554. DOI:10.1039/d0ta11144a |
| [33] |
B. Tian, S. Li, S. Lei, et al., Chinese Chem. Lett. 32 (2021) 2469-2473. DOI:10.1016/j.cclet.2021.02.011 |
| [34] |
Q. Gao, L. Zhang, C. Zheng, et al., Chin. Chem. Lett. (2021) 10.1016/j.cclet.2021.11.027. DOI:10.1016/j.cclet.2021.11.027 |
| [35] |
M. Miao, X. Gong, S. Lei, et al., Chem. Phys. 548 (2021) 111249. DOI:10.1016/j.chemphys.2021.111249 |
| [36] |
Z.W. Chen, J.M. Yan, Q. Jiang, Small Methods 3 (2019) 1800291. DOI:10.1002/smtd.201800291 |
| [37] |
Y. Qian, Y. Liu, Y. Zhao, et al., EcoMat. 2 (2020) e12014. |
| [38] |
X. Guo, J. Gu, S. Lin, et al., J. Am. Chem. Soc. 142 (2020) 5709-5721. DOI:10.1021/jacs.9b13349 |
| [39] |
Y. Ying, X. Luo, J. Qiao, Adv. Funct. Mater. 31 (2021) 2007423. DOI:10.1002/adfm.202007423 |
| [40] |
Z. Wei, J. He, Y. Yang, et al., J. Energy Chem. 53 (2021) 303-308. DOI:10.1016/j.jechem.2020.04.014 |
| [41] |
Y. Li, Q. Zhang, C. Li, et al., J. Mater. Chem. A 7 (2019) 22242-22247. DOI:10.1039/c9ta07845b |
| [42] |
L. Han, Z. Ren, P. Ou, et al., Angew. Chem. Int. Ed. 60 (2021) 345-350. DOI:10.1002/anie.202010159 |
| [43] |
D. Ma, Z. Zeng, L. Liu, et al., J. Phys. Chem. C 123 (2019) 19066-19076. DOI:10.1021/acs.jpcc.9b05250 |
| [44] |
T. Deng, C. Cen, H. Shen, et al., J. Phys. Chem. Lett. 11 (2020) 6320-6329. DOI:10.1021/acs.jpclett.0c01450 |
| [45] |
H. Zhang, C. Cui, Z. Luo, J. Phys. Chem. C 124 (2020) 6260-6266. DOI:10.1021/acs.jpcc.0c00486 |
| [46] |
S. Wang, L. Shi, X. Bai, et al., ACS Cent. Sci. 6 (2020) 1762-1771. DOI:10.1021/acscentsci.0c00552 |
| [47] |
Z. Zhang, X. Xu, ACS Appl. Mater. Interfaces 12 (2020) 56987-56994. DOI:10.1021/acsami.0c16362 |
| [48] |
L. Jasin Arachchige, Y. Xu, Z. Dai, et al., J. Phys. Chem. C 124 (2020) 15295-15301. DOI:10.1021/acs.jpcc.0c03899 |
| [49] |
Z. Shu, Y. Cai, J. Mater. Chem. A 9 (2021) 16056-16064. DOI:10.1039/d1ta03420k |
| [50] |
Z. Zhang, S. Qi, J. Wang, et al., Appl. Surf. Sci. 563 (2021) 150352. DOI:10.1016/j.apsusc.2021.150352 |
| [51] |
B. Wang, S. Huang, L. Yang, et al., J. Phys. Chem. C 125 (2021) 14253-14262. DOI:10.1021/acs.jpcc.1c02397 |
| [52] |
X. Lv, W. Wei, B. Huang, et al., Nano Lett. 21 (2021) 1871-1878. DOI:10.1021/acs.nanolett.0c05080 |
| [53] |
Z. Wei, J. He, Y. Yang, et al., J. Energy Chem. 53 (2021) 303-308. DOI:10.1016/j.jechem.2020.04.014 |
| [54] |
Y. Xu, Z. Cai, P. Du, et al., J. Mater. Chem. A 9 (2021) 8489-8500. DOI:10.1039/d1ta00262g |
| [55] |
D. Ma, Y. Wang, L. Liu, et al., Phys. Chem. Chem. Phys. 23 (2021) 4018-4029. DOI:10.1039/d0cp04843g |
| [56] |
J. Li, C. Cao, J. Hao, et al., Diam. Relat. Mater. 15 (2006) 1593-1600. DOI:10.1016/j.diamond.2006.01.013 |
| [57] |
H. Yuan, Z. Li, X.C. Zeng, et al., J. Phys. Chem. Lett. 11 (2020) 3481-3487. DOI:10.1021/acs.jpclett.0c00676 |
| [58] |
P. Roy, A. Pramanik, P. Sarkar, J. Phys. Chem. Lett. 12 (2021) 2788-2795. DOI:10.1021/acs.jpclett.1c00421 |
| [59] |
Y. Wu, C. He, W. Zhang, ACS Appl. Mater. Interfaces 13 (2021) 47520-47529. DOI:10.1021/acsami.1c11889 |
| [60] |
G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169. DOI:10.1103/PhysRevB.54.11169 |
| [61] |
P.E. Blöchl, Phys. Rev. B 50 (1994) 17953. DOI:10.1103/PhysRevB.50.17953 |
| [62] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865. DOI:10.1103/PhysRevLett.77.3865 |
| [63] |
S. Grimme, J. Antony, S. Ehrlich, et al., J. Chem. Phys. 132 (2010) 154104. DOI:10.1063/1.3382344 |
| [64] |
H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1976) 5188. DOI:10.1103/PhysRevB.13.5188 |
| [65] |
L. Xie, X. Li, X. Wang, et al., J. Phys. Chem. C 123 (2019) 1846-1851. DOI:10.1021/acs.jpcc.8b10521 |
| [66] |
X. Lv, W. Wei, F. Li, et al., Nano Lett. 19 (2019) 6391-6399. DOI:10.1021/acs.nanolett.9b02572 |
| [67] |
H. Niu, Z. Zhang, X. Wang, et al., Adv. Funct. Mater. 31 (2021) 2008533. DOI:10.1002/adfm.202008533 |
| [68] |
L. Shi, Y. Yin, S. Wang, et al., ACS Catal. 10 (2020) 6870-6899. DOI:10.1021/acscatal.0c01081 |
| [69] |
Z. Xue, X. Zhang, J. Qin, et al., Nano Energy 80 (2021) 105527. DOI:10.1016/j.nanoen.2020.105527 |
| [70] |
C. Ling, X. Niu, Q. Li, et al., J. Am. Chem. Soc. 140 (2018) 14161-14168. DOI:10.1021/jacs.8b07472 |
| [71] |
Z.Q. Chu, C. Stampfl, X.M. Duan, J. Phys. Chem. C 123 (2019) 28739-28743. DOI:10.1021/acs.jpcc.9b08169 |
| [72] |
S. Ji, Z. Wang, J. Zhao, J. Mater. Chem. A 7 (2019) 2392-2399. DOI:10.1039/c8ta10497b |