 2022, Vol. 33
2022, Vol. 33 


b Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing 211198, China;
c Institute of Translation Medicine, Shanghai University, Shanghai 200444, China
Globally, aside from nonmelanoma skin cancer, the most prevalent cancer found in women is breast cancer [1]. In 2022, over 287, 850 people will be newly diagnosed with breast cancer, with the estimation of the American Cancer Society [2]. With the growth of aging populations worldwide, these numbers will continue to rise relentlessly. Therefore, it is imperative to develop advanced therapeutics to treat breast cancer effectively.
Breast cancer occurs in four stages, stages Ⅰ-Ⅳ, and can be grouped into three major subtypes depending on the absence or presence of molecular markers for estrogen or progesterone receptors and human epidermal growth factor 2 (ERBB2) [3]. The three major subtypes are as follows: hormone receptor-positive/ERBB2 negative (HR+/ERBB2−), ERBB2 positive (ERBB2+), and triple-negative. Approximately 70%, 15%-20%, and 15% of patients are grouped under HR+/ERBB2−, ERBB2+, and triple-negative, respectively [4-6]. While stage Ⅰ breast cancer have a 5-year breast cancer-specific survival rates for HR+/ERBB2−, ERBB2+, and triple-negative are approximately 99%, 94%, and 85%, respectively, the median overall survival of stage Ⅳ breast cancer is about 5 years for HR+ or ERBB2+ subtypes and 1 year for triple-negative, with the possible recurrence of the triple-negative breast cancer [7-10]. Despite the related long- and short-term risks, the aversion of recurrence and imperative breast cancer treatment relies on chemotherapy.
Daunorubicin (DNR) is an anthracycline and exhibits its antimitotic and cytotoxic activities via multiple mechanisms, including forming complexes with DNA, inhibition of topoisomerase Ⅱ activity, inhibition of DNA polymerase activity, regulation of gene expression, and production of DNA-damaging free radicals [11]. Cytarabine (ara-C) is an antimetabolite and mainly affects the cell cycle by inhibiting DNA polymerase activity [12]. Combining these two anticancer drugs at a molar ratio of 1:5 has been demonstrated to yield maximally synergistic and minimally antagonistic anticancer activities both in vitro and in vivo [13]. The optimized drug combination has been further fixed in nanoscale liposomes, which can deliver the most effective synergistic ratio of drugs to cancer cells after being administrated in a relatively more straightforward manner [13]. However, these liposomes' specific cancer cell targeting capacity could be further improved to maximize their therapeutic potentials.
Cancer cell targetability and site-specific drug release are critical factors that can facilitate liposomes with the capacity for effective eradication of malignancy [14]. It is generally accepted that the maximum therapeutic potential can be achieved by modifying liposomes, which can specifically bind to the receptor and be internalized by endocytosis, then quickly release drugs at the intracellular level [15]. Fortunately, CD44, a receptor known to be overexpressed in patients with cancer [16], offers the possibility of optimizing the liposomal formulation for the specific receptor-binding mediated intracellular co-delivery DNR and ara-C.
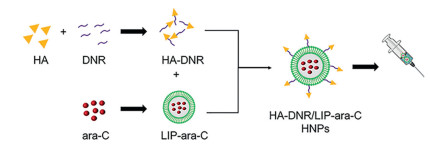
Herein, we have developed a targeted DNR and ara-C co-delivery system based on the hyaluronic acid (HA) modified liposomes. As illustrated in Scheme 1, HA-DNR prodrug was firstly synthesized and then assembled onto the ara-C-loaded liposomes (LIP-ara-C) to form the hybrid nanoparticles (HA-DNR/LIP-ara-C HNPs). Since the HA, a linear polysaccharide composed of d-glucuronic acid and N-acetyl-d-glucosamine, can specifically bind to CD44 [17]. The HA-DNR/LIP-ara-C HNPs were hypothesized to target the CD44 presented on cancer cells specifically, be internalized through endocytosis, and then released drugs intracellularly. The released drug combination could inhibit the intracellular synthesis of DNA and RNA, leading to the apoptosis of cancer cells. Accordingly, the maximum therapeutic potential of HA-DNR/LIP-ara-C HNPs was believed to realize for the targeted and synergistic treatment of AML.

|
Download:
|
| Scheme 1. Schematic illustration of HNP formation. The HNPs were co-assembled from two functional building blocks: HA-DNR prodrug and LIP-ara-C. The HA-DNR prodrug was firstly synthesized and then assembled onto the prepared LIP-ara-C to form spherical HNPs. | |
{kind=link}
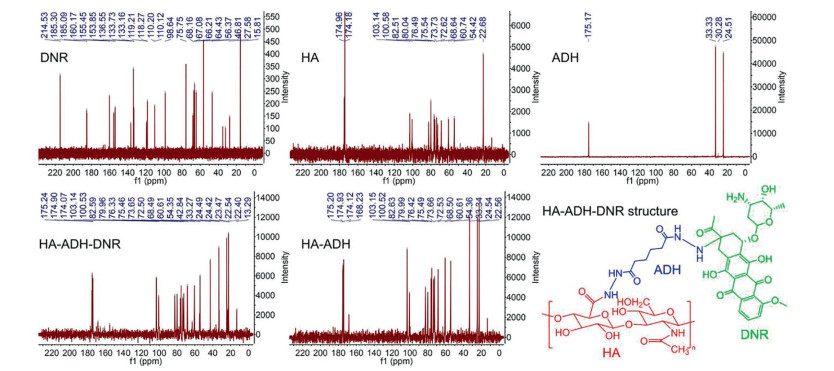
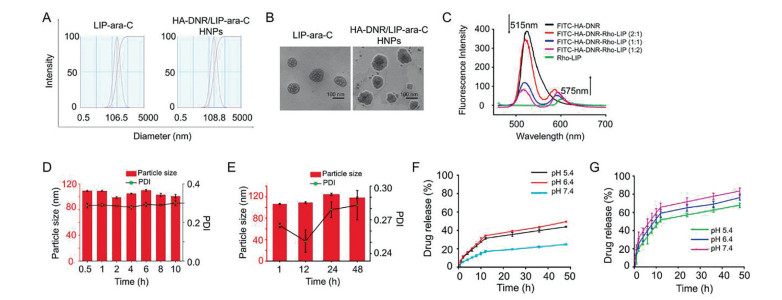
To fabricate HA-DNR/LIP-ara-C HNPs, the two functional building blocks, we first prepared HA-DNR prodrug and LIP-ara-C. The HA-DNR prodrug was synthesized by conjugating HA to DNR via an acid-sensitive hydrazone linkage and had a DNR loading of 2.29% [18]. The successful synthesis was confirmed by 13C nuclear magnetic resonance (13C NMR) spectrometer (Fig. 1). The amide ADH peak at δ 175 transferred to δ 168 in the HA-ADH, and the peak intensity reduced correspondingly in HA-ADH-DNR, owing to the HA or DNR connection. In HA-ADH-DNR, the peaks at δ 175, δ 174, and δ 168 were the characterized peak of amide in HA and ADH, respectively. The special benzene ring peak of DNR also appeared in HA-ADH-DNR at around δ 100, δ 150 and δ 160. Meanwhile, the LIP-ara-C was prepared using the film hydration method followed by extrusion [19]. By adjusting the amount of encapsulated ara-C and condition of ultrasonication, the optimal LIP-ara-C with the highest drug loading was prepared (Fig. S1 in Supporting information). Based on the UV-vis spectrometer, the loading rate and encapsulation efficiency of ara-C in the optimized formulation were calculated to be 13.7% and 23.75%, respectively, whereas the loading rate and encapsulation efficiency of DNR were 0.45% and 7.48%, respectively. The HA-DNR/LIP-ara-C HNPs were formed by mixing the solutions containing the two functional building blocks and vortex. According to our previous work, the HA-based prodrugs have the potential to assemble directly onto liposomes since the hydrophobic interactions could surpass the electrostatic repulsive forces between the negatively charged HA prodrug and liposomes [20]. Thus, the HA-DNR prodrug was assumed to assemble onto the LIP-ara-C by inserting the hydrophobic DNR into the bilayers of the liposome. As a result, the HA-DNR/LIP-ara-C HNPs, with the decoration of HA-DNR prodrug at the surface, have a slightly larger size (Fig. 2A) and higher zeta-potential (Table S1 in Supporting information) than the LIP-ara-C. The spherical morphology of these nanoparticles with a diameter around 100 nm was further demonstrated by TEM (Fig. 2B).

|
Download:
|
| Fig. 1. 13C NMR spectra of hyaluronic acid (HA), adipic acid dihydrazide (ADH), hyaluronic acid- adipic acid dihydrazide conjugate (HA-ADH), daunorubicin (DNR), and hyaluronic acid-daunorubicin prodrug (HA-ADH-DNR or HA-DNR). | |
{kind=link}

|
Download:
|
| Fig. 2. (A) Particle size distribution of LIP-ara-C and HA-DNR/LIP-ara-C HNPs. (B) Representative TEM images of LIP-ara-C and HA-DNR/LIP-ara-C HNPs. The scale bar is 100 nm. (C) Fluorescence emission spectra of FITC-HA-PTX/Rho-LTSL HNPs with FITC/Rho mass ratios of 1:0, 2:1, 1:1, 1:2, and 0:1. The excitation wavelength is at 450 nm. (D) Particle size and PDI of HA-DNR/LIP-ara-C HNPs incubated in 10% PBS at 37 ℃ for 10 h showing high stability in vitro. (E) Particle size and PDI of HA-DNR/LIP-ara-C HNPs incubated in PBS at 4 ℃ for 48 h showing high storage stability in vitro. (F) In vitro release profile of DNR from HA-DNR/LIP-ara-C HNPs in PBS at pH 5.4, 6.4, and 7.4. (G) In vitro release profile of ara-C from HA-DNR/LIP-ara-C HNPs in PBS at pH 5.4, 6.4, and 7.4. mean ± SD, n = 3. | |
{kind=link}
To confirm the successful assembly of the HA-DNR prodrug onto the surface of the negatively charged LIP-ara-C, we utilized fluorescence resonance energy transfer (FRET), an important technique for investigating molecular proximity (typically 10-100 Å) [19]. Specifically, a donor molecular (FITC) with a wavelength of 515 nm was conjugated to the HA-DNR prodrug, while an acceptor molecular (RHO) with a wavelength of 575 nm was loaded into the liposomes. The FITC-HA-DNR/RHO-LIP NPs were then prepared at varying mass ratios (2:1, 1:1, 1:2) and their fluorescence emission spectra were recorded. As shown in Fig. 2C, both FITC-HA-DNR and RHO-LIP emitted fluorescence upon excitation at 450 nm. With the proportion increase of RHO-LIP, the fluorescence peak of FITC (donor) at 515 nm decreased, while the one for RHO (acceptor) at 575 nm increased gradually. These changes in fluorescence intensity indicated FITC-HA-DNR is in close interaction with RHO-LIP so that the excitation from the FITC donor could transfer to the RHO acceptor without emitting photons during the electronically excited states of the two molecules. Accordingly, co-assembly of the HA-DNR prodrug and the LIP-ara-C was proven successful.
For stability evaluation of HA-DNR/LIP-ara-C HNPs, their particle size and polydispersity index (PDI) value were monitored in 10% fetal bovine serum (FBS) or phosphate-buffered saline (PBS). As depicted in Figs. 2D and E and Fig. S2 (Supporting information), no significant differences were observed after incubation in 10% FBS or PBS, suggesting the excellent serum stability and storage stability of HA-DNR/LIP-ara-C HNPs, respectively. In other words, increased circulation half-life should be achieved for HA-DNR/LIP-ara-C HNPs after being administrated in vivo. Moreover, the high stability of HA-DNR/LIP-ara-C HNPs also confirmed that the HA-DNR prodrug co-assembled onto the LIP-ara-C via incorporation of its hydrophobic moiety into the lipid membranes, rather than coating on the surface of HA-DNR/LIP-ara-C HNPs via electrostatic interactions.
The release profile of DNR and ara-C from the HNPs was then evaluated in vitro. Under different pH conditions, the release of both DNR (Fig. 2F) and ara-C (Fig. 2G) reached equilibrium within 24 h. The highest release of DNR was obtained at pH 6.4 (Fig. 2F), whereas the highest release of ara-C was obtained at pH 7.4 (Fig. 2G). These results indicated that both DNR and ara-C could be released at the target site in a sustained manner.
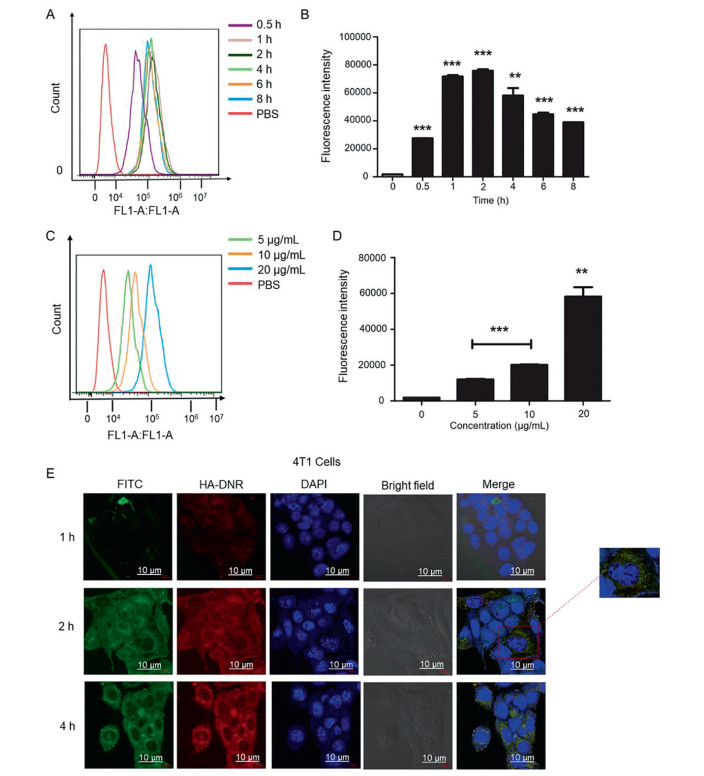
The 4T1 cell line with overexpressed CD44 receptors was used to examine the CD44-mediated intracellular uptake of HNPs. We prepared fluorescence-labeled HNPs (HA-DNR/FITC-LIP HNPs) and incubated them with the 4T1 cells to enable the quantitative and qualitative analysis. When a fixed concentration of HA-DNR/FITC-LIP HNPs (20 µg/mL of FITC) was used for the incubation, the intracellular uptake of HNPs reached equilibrium within 2 h and declined after 4 h (Figs. 3A and B). When an increasing concentration of HA-DNR/FITC-LIP HNPs was used for the incubation, the intracellular uptake of HNPs increased accordingly (Figs. 3C and D). Therefore, based on these quantitative analyses by flow cytometry, the HA-DNR/FITC-LIP HNPs were demonstrated to be efficiently internalized by the 4T1 cells in both time- and concentration-dependent manners. The intracellular uptake of HA-DNR/FITC-LIP HNPs was also qualitatively monitored by confocal laser scanning microscopy (CLSM). As presented in Fig. 3E, the uptake equilibrium at 2 h was observed with strong fluorescence intensity, consistent with the flow cytometry analysis. The green fluorescence of FITC-LIP was then merged with the red fluorescent of HA-DNR and generated a yellow color, signifying the HA-DNR/FITC-LIP HNPs were located in the lysosomes before accumulating in the cytoplasm. Overall, the CD44 mediated intracellular uptake and localization of the HA-DNR/FITC-LIP HNPs was confirmed to occur through an endocytic pathway.

|
Download:
|
| Fig. 3. (A) Time-dependent uptake of HA-DNR/FITC-LIP in 4T1 cells at a fixed FITC concentration of 20 µg/mL measured by flow cytometry. (B) Bar chart for the time-dependent uptake of HA-DNR/FITC-LIP in 4T1 cells at a fixed FITC concentration of 20 µg/mL (mean ± SD, n = 3, **P < 0.01 and ***P < 0.001). (C) Concentration-dependent uptake of HA-DNR/FITC-LIP in 4T1 cells at varying FITC concentrations of 5 µg/mL, 10 µg/mL, and 20 µg/mL measured by flow cytometry. (D) Bar chart for the concentration-dependent uptake of HA-DNR/FITC-LIP in 4T1 cells at varying FITC concentrations of 5 µg/mL, 10 µg/mL, and 20 µg/mL. (mean ± SD, n = 3, **P < 0.01 and ***P < 0.001). (E) CLSM images of 4T1 cells treated with HA-DNR/FITC-LIP for 1 h, 2 h, and 4 h at a fixed FITC concentration of 7.5 µg/mL under 37 ℃. The scale bar is 10 µm. | |
{kind=link}
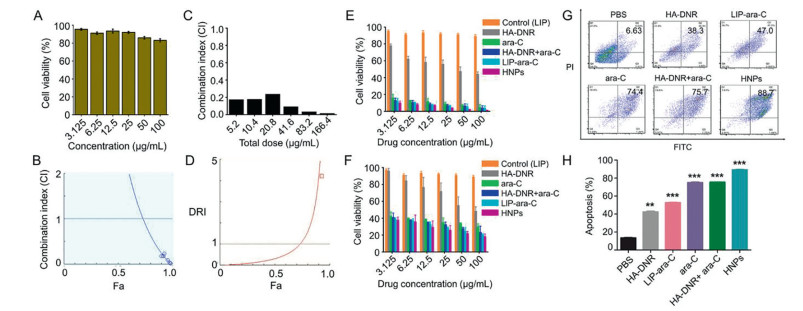
To evaluate the cancer-cell killing efficiency of HA-DNR/LIP-ara-C HNPs, we first examined the cytotoxicity of the drug-free liposomes. As displayed in Fig. 4A, cell viabilities were kept to almost 100% even after being treated with the highest concentration of drug-free liposomes, suggesting the HNPs have no cytotoxicity before accumulating at the target site. The combinational effect of the co-delivered DNR and ara-C, synergistic, additive, or antagonistic, was then determined. Specifically, free DNR and ara-C combinations with varying molar ratios were used to treat the 4T1 cells. Based on their cytotoxicity, the combination index (CI) was calculated. The lowest CI value was obtained from the DNR and ara-C combination at a molar ratio of 1:5 (Fig. S3 in Supporting information), suggesting DNR and ara-C act synergistically on the 4T1 cells. Additionally, the dose reduction index (DRI) of the free drug combinations was also determined to depict how many folds the dose of each drug may be reduced based on synergism, compared with the dosage of each drug alone. Fortunately, the 1:5 DNR and ara-C combination presented the mostly reduced dose-related toxicity compared to the other two combinations (Fig. S4 in Supporting information). On these bases, the CI and DRI values of HA-DNR/LIP-ara-C HNPs were calculated. After being incorporated in the HNPs, the optimized DNR and ara-C combination demonstrated a comparatively more significant synergistic effect (Figs. 4B and C) with a significantly reduced dose-related toxicity (Figs. 4D-F) than its free form.

|
Download:
|
| Fig. 4. (A) Cell viability of 4T1 cells treated with varying concentrations of LIP, ranging from 3.125 µg/mL to 100 µg/mL for 48 h, n = 3. (B) Combination index (CI) for 4T1 cells treated with HA-DNR/LIP-ara-C HNPs of molar ratio 1:5 for 48 h. CI < 1, CI = 1 and CI > 1 indicate synergistic, additive and antagonistic effects, respectively. (C) Bar chart of the combination index (CI) for 4T1 cells treated with HA-DNR/LIP-ara-C HNPs of molar ratio 1:5 for 48 h. (D) Dose reduction index (DRI) for 4T1 cells treated with HA-DNR/LIP-ara-C HNPs of molar ratio 1:5 for 48 h. (E) Cell viability of 4T1 cells after treatment with ara-C concentration ranging from 3.125 µg/mL to 100 µg/mL for 48 h. The mass ratio for free ara-C and HA-DNR combination and HA-DNR/LIP-ara-C HNPs were 1:1.25 (mean ± SD, n = 3). (F) Cell viability of 4T1 cells after treatment with ara-C concentration ranging from 3.125 µg/mL to 100 µg/mL for 24 h. The mass ratio for free ara-C and HA-DNR combination and HA-DNR/LIP-ara-C HNPs were 1:1.25 (mean ± SD, n = 3). (G) A flow cytometry analysis for apoptosis on 4T1 cells after treatment with different formulations at DNR or ara-C concentration of 100 µg/mL for 48 at 37 ℃, n = 3. (H) Quantitative analysis of apoptosis in 4T1 cells. The mass ratio for free ara-C and HA-DNR combination and HA-DNR/LIP-ara-C HNPs were 1:1.25 (mean ± SD, n = 3, **P < 0.01 and ***P < 0.001 as compared to the control). | |
{kind=link}
To explore the cancer cell death mechanism induced by HA-DNR/LIP-ara-C HNPs, we performed Annexin V-FITC/PI staining assay on the 4T1 cells treated with various drugs formulations. Then both apoptotic and necrotic cells were quantified by flow cytometry. Compared to the other formulations, the highest apoptotic rate at 88.7% was observed for cells treated with HA-DNR/LIP-ara-C HNPs, even though the free DNR and ara-C combination-treated cells also exhibited a high apoptotic rate at 75.7% (Figs. 4G and H). Since apoptosis is a highly controlled and regulated cell death process, the inadequate apoptosis typically results in cell proliferation and cancer development [21]. Therefore, the highest apoptotic rate of HA-DNR/LIP-ara-C HNPs indicates its success in cancer-cell killing.
In summary, a targeted DNR and ara-C co-delivery system were developed based on hybrid nanoparticles (HA-DNR/LIP-ara-C HNPs), in which synthesized HA-DNR prodrug were co-assembled onto prepared LIP-ara-C. The HA-DNR/LIP-ara-C HNPs, with particle size at approximately 100 nm, exhibited good serum and storage stability, indicating improved circulation half-time when administrated in vivo. Through targeted binding to the CD44 receptor that overexpressed on 4T1 cancer cells, these HNPs could be internalized by 4T1 cells via endocytosis, enabling enhanced intracellular accumulation. The optimized DNR and ara-C combination at a molar ratio of 1:5 could then be sustained released intracellularly, generating enhanced synergistic effects with reduced dose-related toxicity. Unlike other lipid prodrugs, which are generally absorbed passively to tumor or target sites [22], assembling our prodrug onto the liposomes potentially allows the co-delivery system to actively target tumors at specific sites with increased stability and retention. Even though further pharmacological and pharmacokinetic studies on proper animal models should be done, these promising properties of HA-DNR/LIP-ara-C HNPs envisioned their improved anticancer efficacy than the commercialized liposomal formulation for the treatment of breast cancer in the future.
Declaration of competing interestThe authors report no declarations of competing interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 81872823 and 82073782), the Double First-Class (No. CPU2018PZQ13) of the China Pharmaceutical University, the Shanghai Science and Technology Committee (No. 19430741500), and the Key Laboratory of Modern Chinese Medicine Preparation of Ministry of Education of Jiangxi University of Traditional Chinese Medicine (No. zdsys-202103).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.04.033.
| [1] |
F. Bray, J. Ferlay, I. Soerjomataram, et al., CA Cancer J. Clin. 68 (2018) 394-424. DOI:10.3322/caac.21492 |
| [2] |
R.L. Siegel, K.D. Miller, H.E. Fuchs, A. Jemal, CA Cancer J. Clin. 72 (2022) 7-33. DOI:10.3322/caac.21708 |
| [3] |
A.G. Waks, E.P. Winer, JAMA 321 (2019) 288-300. DOI:10.1001/jama.2018.19323 |
| [4] |
H. Joshi, M.F. Press, 22 - Molecular oncology of breast cancer, in: K.I. Bland, E.M. Copeland, V.S. Klimberg, W.J. Gradishar (Eds. ), The Breast, Elsevier, Amsterdam, 2018, pp. 282-307. e5
|
| [5] |
M. Piccart-Gebhart, M. Procter, B. Leyland-Jones, et al., N. Engl. J. Med. 353 (2005) 1659-1672. DOI:10.1056/NEJMoa052306 |
| [6] |
C. Denkert, C. Liedtke, A. Tutt, G. von Minckwitz, The Lancet 389 (2017) 2430-2442. DOI:10.1016/S0140-6736(16)32454-0 |
| [7] |
M. Chavez-MacGregor, E.A. Mittendorf, C.A. Clarke, et al., Oncologist 22 (2017) 1292. DOI:10.1634/theoncologist.2017-0116 |
| [8] |
A. Bardia, I.A. Mayer, J.R. Diamond, et al., J. Clin. Oncol. 35 (2017) 2141. DOI:10.1200/JCO.2016.70.8297 |
| [9] |
S.M. Swain, J. Baselga, S.B. Kim, et al., N. Engl. J. Med. 372 (2015) 724-734. DOI:10.1056/NEJMoa1413513 |
| [10] |
J.F. Robertson, J.P. Lindemann, A. Llombart-Cussac, et al., Breast Cancer Res. Tr. 136 (2012) 503-511. DOI:10.1007/s10549-012-2192-4 |
| [11] |
B. Löwenberg, G.J. Ossenkoppele, W. van Putten, et al., New Engl. J. Med. 361 (2009) 1235-1248. DOI:10.1056/NEJMoa0901409 |
| [12] |
R.L. Momparler, Exp. Hematol. Oncol. 2 (2013) 1-5. DOI:10.1186/2162-3619-2-1 |
| [13] |
J.E. Lancet, G.L. Uy, J.E. Cortes, et al., J. Clin. Oncol. 36 (2018) 2684. DOI:10.1200/jco.2017.77.6112 |
| [14] |
W. Wu, Y. Lu, J. Qi, Ther. Deliv. 6 (2015) 1239-1241. DOI:10.4155/tde.15.69 |
| [15] |
W. He, X. Xing, X. Wang, et al., Adv. Funct. Mater. 30 (2020) 1910566. DOI:10.1002/adfm.201910566 |
| [16] |
R. Quere, S. Andradottir, A. Brun, et al., Leukemia 25 (2011) 515-526. DOI:10.1038/leu.2010.281 |
| [17] |
W. Li, C. Zhou, Y. Fu, et al., Acta Pharma. Sin. B 10 (2020) 693-710. DOI:10.1016/j.apsb.2019.07.003 |
| [18] |
Y. Luo, G.D. Prestwich, Bioconjugate Chem. 10 (1999) 755-763. DOI:10.1021/bc9900338 |
| [19] |
W. Liu, D. Li, Z. Dong, et al., Int. J. Pharm. 587 (2020) 119682. DOI:10.1016/j.ijpharm.2020.119682 |
| [20] |
Y. Lv, C. Xu, X. Zhao, et al., ACS Nano 12 (2018) 1519-1536. DOI:10.1021/acsnano.7b08051 |
| [21] |
B.A. Carneiro, W.S. El-Deiry, Nat. Rev. Clin. Oncol. 17 (2020) 395-417. DOI:10.1038/s41571-020-0341-y |
| [22] |
A. Arouri, A.H. Hansen, T.E. Rasmussen, O.G. Mouritsen, Curr. Opin. Colloid Interface Sci. 18 (2013) 419-431. DOI:10.1016/j.cocis.2013.06.001 |