 2022, Vol. 33
2022, Vol. 33 


b School of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, China
Ever since the discovery of the analgesic effects of opioids more than a century ago, they are still the mainstay of the treatment for moderate-to-severe pain. Unfortunately, the adverse effects of the present opioid analgesics, such as constipation, physical dependence, tolerance, respiratory depression, and the current opioid epidemic, limit their clinical application [1-4]. Over-reliance on fentanyl derivatives also has led to the recent opioid crisis. To date, there are no effective approaches to address this high unmet medical need. Therefore, efforts to develop safer opioids should be continued.
Opioid analgesics exert pharmacological effects by interacting with specific opioid receptors, including µ-opioid receptor (MOR), δ-opioid receptor (DOR), and κ-opioid receptor (KOR). The initial mainstream opioid analgesics developed included mono-target-directed ligands with a potent activity and a high selectivity. However, many literature reports indicate that these potent mono-target-directed ligands have their own limitations due to adverse effects. Therefore, potent mono-target-directed ligands cannot meet current clinical needs [5, 6]. The synergistic antinociceptive effect between MOR agonists and DOR agonists was found in recent years and could be used as a new strategy for the discovery of safer opioids [7-12].
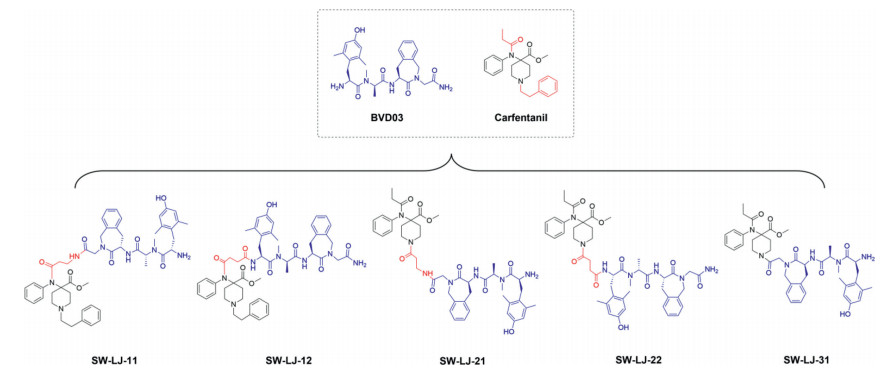
Herein, novel opioid peptide–fentanyl analogue conjugates were synthesized as dual µ/δ-opioid receptor (MOR/DOR) agonists to obtain new opioid analgesics with diminished deleterious effects. By the covalent coupling of derivatives of carfentanyl, an ultra-potent synthetic opioid fentanyl analogue with high specificity for the MOR, to the chemically modified endogenous opioid peptide BVD03, a dermorphin tetrapeptide with mixed MOR/DOR activity, hybrid conjugates as synergistic bifunctional ligands were generated (Fig. 1) [13-16]. By the conjugating of fentanyl analogue to the BVD03 peptide, the MOR agonist activity of BVD03 was improved and the adverse effects of fentanyl compound might be diminished due to the potent DOR activity of BVD03. These new conjugates were evaluated in vitro for their affinity and activity on both the MOR and DOR as well as tested in vivo for their analgesic efficacy in four classic animal models of pain. The most potent conjugate was further assessed for its effects on physical dependence and respiratory depression.

|
Download:
|
| Fig. 1. The chemical structures of BVD03–fentanyl analogue conjugates. | |
BVD03 was prepared by liquid-phase peptide synthesis, and fentanyl derivatives were coupled to the N-terminus or C-terminus of BVD03 via a chemical linker to obtain the novel conjugates SW-LJ-11, SW-LJ-12, SW-LJ-21, SW-LJ-22 and SW-LJ-31 (Schemes S1–S3 in Supporting information). The purities of all test compounds by high performance liquid chromatography (HPLC) were > 98.0%.
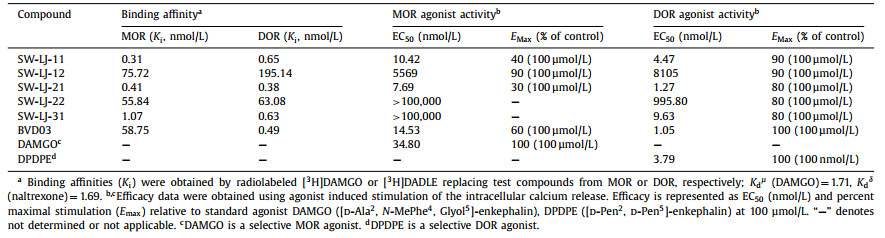
The in vitro binding affinity and functional activity at the MOR and DOR were established for BVD03 and the hybrid compounds (Table 1). All conjugates maintained affinities within the moderate-to-low nanomolar range at both the MOR and DOR. SW-LJ-11, SW-LJ-21, and SW-LJ-31 displayed a significantly enhanced binding affinity (nearly 55–190-fold) to the MOR compared with BVD03. Surprisingly, the functional potency of the conjugates did not always reflect the binding affinity of the compounds. SW-LJ-11 and SW-LJ-21 displayed potent MOR/DOR agonist activity in the low nanomolar range as a partial agonist for the MOR and as a full agonist for the DOR, while the other conjugates were found to be devoid of significant agonist efficacy at the MOR. Because SW-LJ-22 and SW-LJ-31 displayed strong-to-moderate MOR binding affinity but low agonist activity, they likely act as MOR antagonists. However, this was not explicitly assayed.
|
|
Table 1 Binding affinity and activity at µ/δ-opioid receptors. |
The antinociceptive potency and efficacy of the conjugates were evaluated in mice using a formalin injection nociception assay. All the procedures adopted were approved and agreed upon by the Institutional Animal Ethical Committee of Beijing Institute of Pharmacology and Toxicology. The research was conducted in accordance with the ethical standards. All care and use guidelines for laboratory animals were followed.
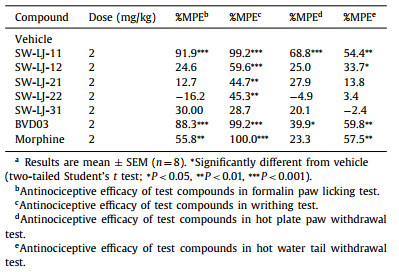
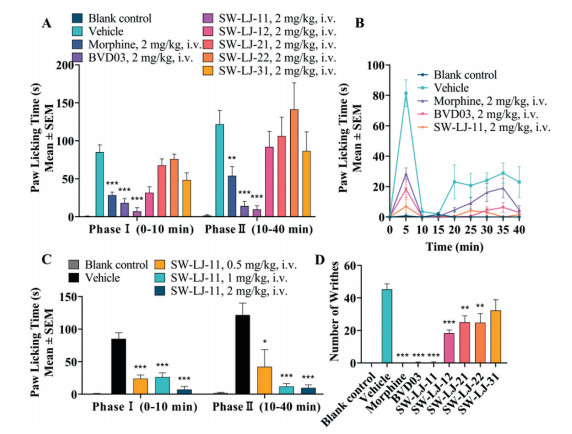
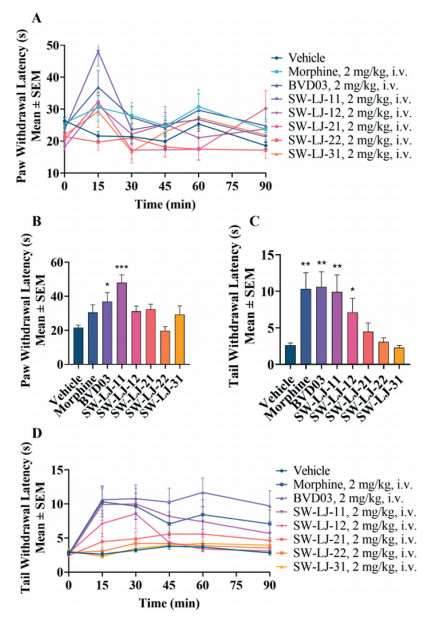
The test compounds, BVD03 and morphine, were injected via the tail vein of mice at a dosage of 2 mg/kg, respectively. As shown in Figs. 2A and B, and Table 2, SW-LJ-11 and BVD03 produced sustained analgesia in phase Ⅰ and phase Ⅱ of the formalin paw licking test (P < 0.001; compared with the vehicle group), with a percent maximum possible effect (%MPE) of 91.9 and 88.3 in phase Ⅱ, respectively. By comparison, morphine also showed sustained analgesia and less potency than SW-LJ-11 and BVD03, with a %MPE of 55.8 at the same dosage. Furthermore, SW-LJ-11 produced dose-dependent anti-nociception following tail vein administration of 0.5, 1.0 and 2.0 mg/kg doses (Fig. 2C).

|
Download:
|
| Fig. 2. The test compounds produced antinociceptive behavior in an acute pathological pain model assay. The results are expressed as the mean ± standard error of mean (SEM) (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle (two-tailed Student's t-test). The blank control consisted of mice that were treated with saline i.v. and injected with saline i.paw. (in the formalin paw licking test) and i.p. (in the writhing test). (A) The test compounds decreased the nociceptive behavior in the formalin paw licking test. (B) The time course of antinociception of morphine, BVD03, and SW-LJ-11 in the formalin paw licking test. (C) The analgesic activity of SW-LJ-11 after i.v. injection at three different doses in the formalin paw licking test. (D) Antinociception of the test compounds (2 mg/kg, i.v.) in the writhing test. | |
|
|
Table 2 Antinociceptive efficacy of the test compounds in vivo.a |
{kind=link}
{kind=link}
The analgesic effects of the novel conjugates in the writhing test are shown in Fig. 2D and Table 2. Consistent with their formalin assay activities, SW-LJ-11, BVD03, and morphine exhibited potent analgesia in the writhing test, with a %MPE of up to 99.2 at the same dosage of 2 mg/kg injected via the tail vein of mice. The other conjugates showed less potent analgesia compared to SW-LJ-11, BVD03, and morphine.
The mouse hot plate paw withdrawal test and the warm water tail-flick assay were used as acute physiological pain models to investigate the antinociceptive effects of the novel compounds. The analgesia in the mouse hot plate paw withdrawal test is shown in Figs. 3A and B, and Table 2. SW-LJ-11 exhibited the most significant analgesic potency in the hot plate test at the time of the peak analgesic effect, with a %MPE of 68.8. This %MPE is 1.7 times greater than that of BVD03 at the same dosage of 2.0 mg/kg. Meanwhile, morphine and the other test compounds showed no significant analgesic effects at the same dosage. The analgesia in the mouse warm water tail-flick assay is shown in Figs. 3C and D, and Table 2. At 15 min following administration, BVD03, morphine, and SW-LJ-11 showed similar %MPE values (59.8, 57.5, and 54.4, respectively), while SW-LJ-11 inhibited pain with the median effective dose (ED50) value of 1.22 mg/kg (Fig. S2 in Supporting information).

|
Download:
|
| Fig. 3. The test compounds produced antinociceptive behavior in an acute physiological pain model assay. The results are expressed as the mean ± SEM (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle (two-tailed Student's t-test). (A) The time course of antinociception of the test compounds in the hot plate paw withdrawal test. (B) Antinociception of the test compounds (2 mg/kg, i.v.) in the hot plate paw withdrawal test at the time of the peak analgesic effect. (C) Antinociception of the test compounds (2 mg/kg, i.v.) in the hot water tail withdrawal test at the time of the peak analgesic effect. (D) The time course of antinociception of the test compounds in the hot water tail withdrawal test. | |
{kind=link}
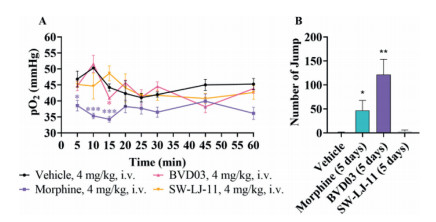
Respiratory depression, a severe adverse effect produced by fentanyl analogues [13], was assessed using blood gas analyser. As depicted in Fig. 4A, the mice receiving SW-LJ-11 did not show a reduction in any respiratory measures across the duration of the experiment, while the morphine-exposed mice did present with a significant reduction in pO2 (mmHg) of blood as compared to the saline control. These data suggest that SW-LJ-11 at the same dose as BVD03 and morphine does not induce respiratory depression acutely. In addition, physical dependence was determined using a precipitated withdrawal approach [17, 18]. Mice were treated with naloxone (1 mg/kg, s.c.) after the administration of the test compounds for five successive days to assess the physical dependence induced by morphine, BVD03, and SW-LJ-11, respectively (Fig. 4B). The mice dosed with morphine or BVD03 jumped significantly more than those treated with the vehicle or SW-LJ-11, suggesting that both morphine and BVD03 induced significant physical dependence and withdrawal compared to the vehicle. In contrast, SW-LJ-11 did not elicit a significant increase when compared with vehicle treatment, indicating that SW-LJ-11 did not induce physical dependence or withdrawal.

|
Download:
|
| Fig. 4. The adverse reactions studied with BVD03 and SW-LJ-11. The results are expressed as the mean ± SEM (n = 5–8). (A) Respiratory inhibition: Groups of mice (n = 5) were administered (4 mg/kg, ~EDmax, i.v.) with morphine, BVD03, or SW-LJ-11. pO2 of blood sampled from mice eyes at various time points were measured. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle (two-tailed Student's t-test). (B) Physical dependence: Groups of mice (n = 8) were dosed (4 mg/kg, ~EDmax, i.v.) once a day (8:00 am) with morphine, BVD03 or SW-LJ-11 for 5 days. On Day 5, the mice were treated with naloxone (1 mg/kg, s.c.) at 2 h after the administration of morphine, BVD03 or SW-LJ-11. The number of jumps was counted over a 20 min period after the injection of naloxone. *Significantly different from the vehicle (two-tailed Student's t-test, **P < 0.01). | |
{kind=link}
In summary, the synthesis and pharmacological activity of new opioid peptide–fentanyl analogue conjugates as potent dual ligands for the MOR and DOR are reported herein. First, we measured the effects of the novel conjugates on the MOR and DOR in vitro. The results showed that the various carfentanyl-related analogues could maintain affinities at both the MOR and DOR, while only SW-LJ-11 and SW-LJ-21, with carfentanyl-related molecules linked to the C-terminus of BVD03, were both highly potent MOR and DOR agonists. The concentration for 50% of maximal effect (EC50) values of SW-LJ-11 and SW-LJ-21 for MOR agonist activity were 10.42 nmol/L and 7.69 nmol/L, respectively, and for DOR agonist activity, they were 4.47 nmol/L and 1.27 nmol/L, respectively. Molecular docking studies using the X-ray crystal structures of the MOR and DOR were also performed to gain insights into the structure–activity relationships (Fig. S1 in Supporting information). The docking pose of SW-LJ-11 showed that it could specifically interact with the key residues in the conformational switch site needed for activation of the receptors. Meanwhile, the compounds SW-LJ-12 and SW-LJ-22, with the same carfentanyl-related chemical moiety linked to the N-terminus of BVD03, exhibited a decrease in potency at the MOR of approximately 500–10, 000-fold and a decrease in potency at the DOR of approximately 1000–2000, compared to SW-LJ-11 and SW-LJ-21, respectively.
Next, the systemic antinociceptive effects of the novel conjugates were evaluated in vivo using four different classic pain mouse models. Only one compound, SW-LJ-11, showed significant analgesic efficacy in all four animal models of pain, including the formalin injection assay, the writhing test, the hotplate test, and the warm water tail-flick assay; the potency of SW-LJ-11 was equal to or greater than that of morphine at the same dosage. Another conjugate, SW-LJ-21, displayed an equally potent binding affinity and activity as SW-LJ-11 in vitro, but it only showed a weak potency or no activity in some mouse model assays. We then estimated the antinociceptive mechanisms of SW-LJ-11 in vivo. With pretreatment of naloxone, naltrindole, or β-funaltrexamine, specific antagonists for the MOR and DOR, the analgesic activity of SW-LJ-11 was abolished as shown in Fig. S3 (Supporting information), suggesting that the antinociceptive effects of this compound are mediated via MOR/DOR. Surprisingly, SW-LJ-11 exhibited no observed physical dependence or respiratory depression, which are severe adverse events associated with fentanyl opioids, suggesting that the use of peptide–small molecule conjugates as dual MOR/DOR agonists may be a promising strategy for obtaining potent and safer opioid analgesics.
In conclusion, the major challenge in the discovery of safer opioids is to develop new compounds with low abuse potential. Efforts have been and continue to focus on developing new approaches to overcome the adverse effects of opioids. Dual-acting or multifunctional ligands design such as the simultaneous targeting of MOR/DOR is a feasible and efficient strategy for the development of novel opioid analgesics. In this paper, novel opioid peptide-fentanyl analogue conjugates as dual MOR/DOR agonists are reported. The conjugate SW-LJ-11 displayed a high binding affinity at the MOR and DOR as well as strong antinociceptive activity in assays performed in vivo. Of significant interest is our finding that unlike morphine and fentanyl opioids, SW-LJ-11 produced no apparent respiratory depression or physical dependence, indicating a useful avenue for finding safer fentanyl opioids.
Declaration of competing interestThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary dataSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.036.
| [1] |
S. Obeng, T. Hiranita, F. León, L.R. McMahon, C.R. McCurdy, J. Med. Chem. 64 (2021) 6523-6548. DOI:10.1021/acs.jmedchem.1c00028 |
| [2] |
S.M. Burns, C.W. Cunningham, S.L. Mercer, ACS Chem. Neurosci. 9 (2018) 2428-2437. DOI:10.1021/acschemneuro.8b00174 |
| [3] |
A.L. Devereaux, S.L. Mercer, C.W. Cunningham, ACS Chem. Neurosci. 9 (2018) 2395-2407. DOI:10.1021/acschemneuro.8b00150 |
| [4] |
N.E. Hagemeier, Am. J. Manag. Care. 24 (2018) S200-s206. |
| [5] |
C.W. Cunningham, W.M. Elballa, S.U. Vold, Neuropharmacology 151 (2019) 195-207. DOI:10.1016/j.neuropharm.2019.03.006 |
| [6] |
P.W. Schiller, Life Sci. 86 (2010) 598-603. DOI:10.1016/j.lfs.2009.02.025 |
| [7] |
Y.S. Lee, V. Kulkarani, S.M. Cowell, et al., J. Med. Chem. 54 (2011) 382-386. DOI:10.1021/jm100982d |
| [8] |
A.A.H. Azzam, J. McDonald, D.G. Lambert, Br. J. Anaesth. 122 (2019) e136-e145. DOI:10.1016/j.bja.2019.03.006 |
| [9] |
J. Starnowska-Sokół, B. Przewłocka, Molecules 25 (2020) 5520. DOI:10.3390/molecules25235520 |
| [10] |
A. Faouzi, R. Uprety, I. Gomes, et al., J. Med. Chem. 63 (2020) 13618-13637. DOI:10.1021/acs.jmedchem.0c00901 |
| [11] |
W. Lei, R.H. Vekariya, S. Ananthan, J.M. Streicher, J. Pain 21 (2020) 146-160. DOI:10.1016/j.jpain.2019.05.017 |
| [12] |
A. Piotrowska, J. Starnowska-Sokół, W. Makuch, et al., Pain 162 (2021) 432-445. DOI:10.1097/j.pain.0000000000002045 |
| [13] |
M.G. Feasel, A. Wohlfarth, J.M. Nilles, et al., AAPS J. 18 (2016) 1489-1499. DOI:10.1208/s12248-016-9963-5 |
| [14] |
B. Vandormael, D.D. Fourla, A. Gramowski-Voss, et al., J. Med. Chem. 54 (2011) 7848-7859. DOI:10.1021/jm200894e |
| [15] |
R.R. Petrov, R.S. Vardanyan, Y.S. Lee, et al., Bioorg. Med. Chem. Lett. 16 (2006) 4946-4950. DOI:10.1016/j.bmcl.2006.06.040 |
| [16] |
Y.S. Lee, R. Petrov, C.K. Park, et al., J. Med. Chem. 50 (2007) 5528-5532. DOI:10.1021/jm061465o |
| [17] |
J.K. Nuamah, F. Sasangohar, M. Erraguntla, R.K. Mehta, BMC Med. Inform. Decis. Mak. 19 (2019) 113. DOI:10.1186/s12911-019-0834-8 |
| [18] |
A.B. Srivastava, J.J. Mariani, F.R. Levin, Lancet 395 (2020) 1938-1948. DOI:10.1016/S0140-6736(20)30852-7 |