 2022, Vol. 33
2022, Vol. 33 


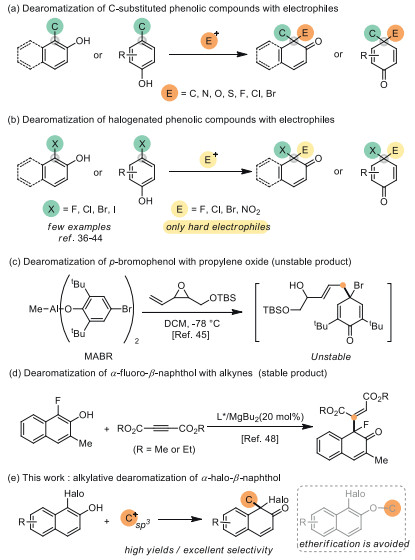
Dearomatization reaction has become a powerful synthetic strategy for the construction of three-dimensional cyclic enones from two-dimensional planar aromatic molecules. In recent years, great efforts have been dedicated to innovations and developments as well as remarkable achievements related to this area [1-5]. For the synthesis of several medicinally important and biological natural products, oxidative dearomatization was recognized as an efficient pathway involving catalytic oxidation of phenols [6-11]. However, the oxidative procedure usually contains difficulties for tolerating sensitive functional groups that would be hard to survive in the presence of oxidants. To overcome this limitation, electrophilic dearomatization using phenols as carbon nucleophiles attracts increasing attention. In this aspect, a series of transformations for C-substituted phenolic molecules have been successfully achieved with different electrophiles (Scheme 1a) [12-35]. Usually, the carbon substituent bonded on the aromatic ring of phenol plays an important role in electrophilic dearomative transformation to enhance the nucleophilicity of the substituted position. In stark contrast, compared with C-substituted phenolic compounds, halophenols generally have a lower nucleophilicity. Therefore, efficient electrophilic dearomatization of halophenols is more challenging than the similar transformation using C-substituted phenols.

|
Download:
|
| Scheme 1. Dearomatization of halophenols. | |
The former electrophilic dearomatization of halophenols showed that the scope of the reaction was mostly limited to hard electrophiles such as halogen- or nitro-electrophiles (Scheme 1b) [36-44]. When soft carbon-electrophiles were applied in such a process, etherification caused by the phenolic hydroxyl group would become an unavoidable problem, and successful examples are few and far between [45-48].
Riccardis and co-workers used AlⅢ-activated halophenols complexes and vinyl epoxides to approach the corresponding dearomatization product, but due to the C(sp3)-Br bonds of dearomatized adducts were unstable, only 1H NMR spectra of the product could be confirmed (Scheme 1c) [45]. Recently, a MgⅡ-catalyzed dearomatization of α-fluoro-β-naphthol with alkynes has been demonstrated, which was the only example for obtaining stable cyclic α-haloenones products through electrophilic dearomatization of halophenols (Scheme 1d) [48]. Besides the above results, the C(sp3)-electrophiles were absent from the direct dearomatization of halophenols. Obviously, compared with electrophilic alkylative dearomatization of halophenols, halogenated dearomatization of alkylphenols could avoid the etherification byproducts and control the regioselectivity very well, and this is the reason why most synthetic methods chose the latter to prepare cyclic α-haloenones from phenolic compounds [48-50]. On the other hand, if a direct and concise alkylation-dearomatization of halophenols could be established, abundant commercial C(sp3)-electrophiles and easily-obtained halophenols would promote this strategy to access new chemical space with the broad application potential. Thus, to overcome the reactive limitations intrinsic to conventional electrophilic alkylative dearomatizations, we designed a direct dearomatization of halonaphthols with C(sp3)-electrophiles (Scheme 1e).
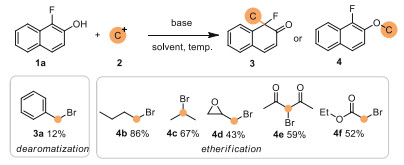
To probe this hypothesis, our exploration was started by testing the reaction performance of α-fluoro-β-naphthol (1a) with various commercially available C(sp3)-electrophiles (2). The preliminary study revealed that alkyl-, isopropyl-, tert-butyl- and electron-deficient alkyl-electrophiles were prone to be attacked by the hydroxyl group of 1a, leading to etherification products 4 without forming any desired product, and only benzyl bromide could participate well in the dearomative pathway with 1a, and the corresponding dearomatized product 3a was successfully detected and isolated in 12% yield (Scheme 2).

|
Download:
|
| Scheme 2. Reaction exploration by combining α-fluoro-β-naphthol (1a) with C(sp3)-electrophiles (2). | |
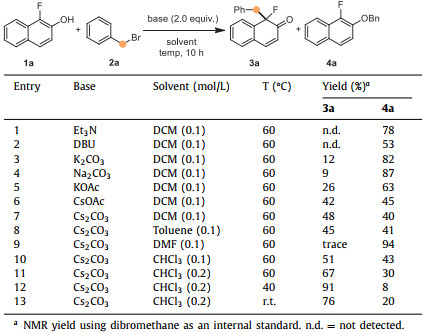
Afterward, α-fluoro-β-naphthol (1a) and benzyl bromide (2a) were used as the model substrates to examine different bases and solvents. The results were summarized in Table 1. Initially, organic bases such as Et3N and DBU were examined in the reaction (Table 1, entries 1 and 2). Unfortunately, only etherification product 4a could be detected. Then we turned our attention to inorganic bases. Low efficiency of the desired dearomatized product was exhibited when K2CO3 and Na2CO3 were used (Table 1, entries 3 and 4). KOAc and CsOAc gave slightly higher yields (Table 1, entries 5 and 6), but 4a caused by etherification was still the major product in the reaction. Fortunately, Cs2CO3 led to better chemoselectivity and promoted the dearomatized pathway to obtain desired product 3a in 48% yield (Table 1, entry 7). Polar aprotic solvent, such as DMF, gave a trace amount of 3a and 94% of etherification byproduct (Table 1, entry 9). Non-polar solvent CHCl3 gave the highest yield compared with DCM and toluene (Table 1, entry 10). Notably, the concentration of reaction mixture had a significant effect on the formation of products. The reaction performed in 0.2 mol/L of CHCl3 led to a rise of desired product 3a to 67% yield (Table 1, entry 11). Decreasing reaction temperature from 60 ℃ to 40 ℃ also promoted the transformation with a higher yield of 91% (Table 1, entry 12). However, the reaction at room temperature gave more O-benzylation byproduct (Table 1, entry 13).
|
|
Table 1 Optimization of the reaction conditions. |
{kind=link}
{kind=link}
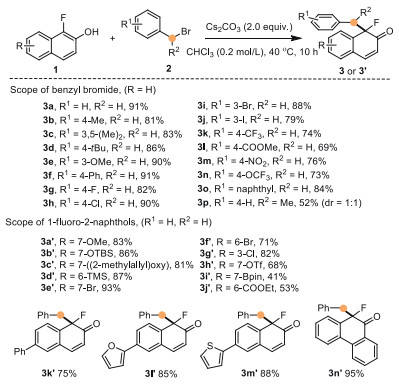
Having identified the optimized dearomatization conditions, the substrate scope was examined (Scheme 3). Benzyl bromides bearing a range of electron-donating and -withdrawing moieties were probed the viability in the reaction process with 1a. Corresponding alkylation dearomatized products (3b-3p) were successfully obtained with moderate to good yields (52%−91%). Not only the electrical neutrality group but also electron-donating substituents bonded on the phenyl rings of benzyl bromides could be well tolerated. Substrates containing electrical neutrality substituents and electron-donating groups, such as methyl (3b, 3c), tert-butyl (3d), methoxyl (3e), and phenyl (3f), reacted well with 1a and converted to desired products in high yields (81%−91%). Moreover, when fluoro (3g), chloro (3h), bromo (3i), and iodo (3j) substituted benzyl bromides were tested, the dearomatization reactions also gave products in good yields (79%−90%). Furthermore, strong electron-withdrawing groups, including trifluoromethyl (3k), methyl ester (3l), nitro (3m), and oxytrifluoromethyl (3n), were tolerated well in the new C-C bond formation leading to the products in moderate to good yields (69%−76%). And 2-(bromomethyl)naphthalene (3o) was also proved as an effective electrophile here. In addition, sterically hindered (1-bromoethyl)benzene could also participate in the dearomatization process to afford product 3p (52% yield, dr =1:1).

|
Download:
|
| Scheme 3. Scope of α-fluoro-β-naphthols with benzyl bromides. | |
{kind=link}
Having established the dearomatization reactions of α-fluoro-β-naphthol with various benzyl bromides, we next aimed to intercept the benzyl electrophile with various fluoronaphthols derivatives to form three-dimensional cyclic enones. A broad range of substituted α-fluoro-β-naphthols (1b-1o) was applied in alkylative dearomatizations with 2a. Again, electron-donating substituents and halide (OMe, OTBS, (2-methylallyl)oxy, TMS, Br, Cl) were well-tolerated and gave the corresponding products in high yields (3a'−3g', 71%–93%). Moreover, α-fluoro-β-naphthols bearing electron-withdrawing substituents (OTf, Bpin, COOEt) gave the desired products successfully, albeit in diminished yields relative to other electron-rich analogs (3h' 68% yield 3i' 41% yield, 3j' 53% yield). We supposed this was because of the detectable increased constructions of O-alkylation byproducts in these transformations. Additionally, aromatic ring and heterocycles (3k' 75% yield, 3l' 85% yield, 3m' 88% yield) were compatible very well in dearomatizations. Notably, phenanthrol substrate gave product 3n' in the yield of 95%.
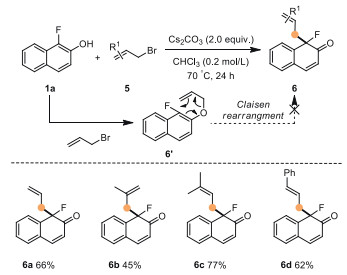
To extend the generality of this method, allyl bromide 5a was tested as another C(sp3)-electrophile (Scheme 4). To our delight, 5a also underwent dearomatization smoothly to afford product 6a in 66% yield. A control experiment for the mechanistic insights on the formation of 6 was carried out with treatment O-allylation adduct 6′ under standard conditions, and no expected product could be detected, indicating that product 6 was not formed through a Claisen rearrangement process. Encouraged by this result, we further looked into the scope of allyl bromides (5b-5d). For substrate 5b, the desired product 6b was obtained in 45% yield. When employing prenyl bromide 5c and cinnamyl bromide 5d, each substrate delivered the desired product in good yield with excellent selectivity for dearomatization over etherification (62%−77%).

|
Download:
|
| Scheme 4. Dearomatization of α-fluoro-β-naphthol with allyl bromides. | |
{kind=link}
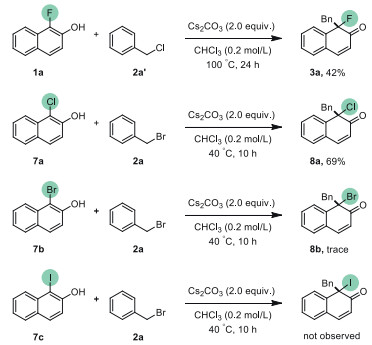
Then, we studied the behavior of benzyl halides with different halogenated naphthols. Using benzyl chloride (2a') instead of benzyl bromide in the dearomatization could also afford 3a in 42% yield. For other halogenated naphthols (X = Cl, Br, I), unprecedented reactivity for α-chloro-β-naphthols (7a) was observed with giving corresponding product 8a in 69% yield (Scheme 5). Only a trace amount of product 8b could be detected when using α-bromo-β-naphthol, and no desired product could be obtained with iodonaphthol 7c.

|
Download:
|
| Scheme 5. Reaction performance of benzyl halide with halogenated naphthols. | |
{kind=link}
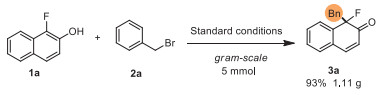
A scaled-up experiment was performed to test the synthetic value of this new method, and 3a (1.11 g) was isolated in 93% yield in the gram-scale synthesis (Scheme 6). Next, the dearomatized product 3a was used for further synthetic transformations (Scheme 7). 3a was treated with MeMgBr in THF at −78 ℃ to obtain tertiary alcohol 9 in 88% yield (dr > 20:1). When H2O2 was used as the oxidant under basic conditions, epoxidation occurred with 3a to afford 10 in 74% yield. Furthermore, oxidizing 3a with m-chloroperbenzoic acid (mCPBA) predominantly provided Baeyer-Villiger product 11 in 48% yield. Exposed 11 to acidic conditions in MeOH leading the ring-opening product 12 in good yield. Then, reactions for the further dearomatization of 3b' were performed. After deprotection with TBAF in the condition of THF, 3b' was converted to a less electron-rich phenol 13, which could have more potential synthetic applications.

|
Download:
|
| Scheme 6. Gram-scale experiments. | |
{kind=link}

|
Download:
|
| Scheme 7. Further synthetic transformations. | |
{kind=link}
In summary, we have achieved the first intermolecular direct dearomatization of halonaphthols with benzyl- and allyl-electrophiles in presence of stoichiometric bases. The three-dimensional cyclic enone products were obtained in high yields with excellent chemoselectivity. Studies on the reaction mechanism revealed the reaction may go through a direct SN2 pathway. Versatile derivatizations of the dearomatization product were proved, which highlighted the potential utility of this concise dearomatization with C(sp3)-electrophiles in further synthetic transformations.
Declaration of competing interestThe authors have declared no conflict of interest.
AcknowledgmentsThe National Natural Science Foundation of China (Nos. 21901203, 22171225, 21925108) and the Education Department of Shaanxi Province (No. 20JK0934) are acknowledged for financial support.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.10.037.
| [1] |
S.P. Roche, J.A. Porco, Angew. Chem. Int. Ed. 50 (2011) 4068-4093. DOI:10.1002/anie.201006017 |
| [2] |
C.X. Zhuo, W. Zhang, S.L. You, Angew. Chem. Int. Ed. 51 (2012) 12662-12686. DOI:10.1002/anie.201204822 |
| [3] |
W.T. Wu, L. Zhang, S.L. You, Chem. Soc. Rev. 45 (2016) 1570-1580. DOI:10.1039/C5CS00356C |
| [4] |
R.X. Liang, L.J. Song, J.B. Lu, et al., Angew. Chem. Int. Ed. 60 (2021) 7412-7417. DOI:10.1002/anie.202014796 |
| [5] |
F.T. Sheng, J.Y. Wang, W. Tan, Y.C. Zhang, F. Shi, Org. Chem. Front. 7 (2020) 3967-3998. DOI:10.1039/D0QO01124J |
| [6] |
S. Dong, J. Zhu, J.A. Porco, J. Am. Chem. Soc. 130 (2008) 2738-2739. DOI:10.1021/ja711018z |
| [7] |
A. Rudolph, P.H. Bos, A. Meetsma, A.J. Minnaard, B.L. Feringa, Angew. Chem. Int. Ed. 50 (2011) 5834-5838. DOI:10.1002/anie.201102069 |
| [8] |
T. Oguma, T. Katsuki, J. Am. Chem. Soc. 134 (2012) 20017-20020. DOI:10.1021/ja310203c |
| [9] |
T. Dohi, N. Takenaga, T. Nakae, et al., J. Am. Chem. Soc. 135 (2013) 4558-4566. DOI:10.1021/ja401074u |
| [10] |
S. Quideau, Modern Arene Chemistry, Wiley-VCH, Weinheim (2002). |
| [11] |
S.K. Jackson, K.L. Wu, R.R. Pettus, Biomimetic Organic Synthesis, Wiley-VCH, Weinheim (2011). |
| [12] |
T. Nemoto, Y. Ishige, M. Yoshida, et al., Org. Lett. 12 (2010) 5020-5023. DOI:10.1021/ol102190s |
| [13] |
Q.F. Wu, W.B. Liu, C.X. Zhuo, et al., Angew. Chem. Int. Ed. 50 (2011) 4455-4458. DOI:10.1002/anie.201100206 |
| [14] |
C.X. Zhuo, S.L. You, Angew. Chem. Int. Ed. 52 (2013) 10056-10059. DOI:10.1002/anie.201304591 |
| [15] |
L. Shao, X. Hu, Chem. Commun. 53 (2017) 8192-8195. DOI:10.1039/C7CC03034G |
| [16] |
Q. Guo, M. Wang, H. Liu, R. Wang, Z. Xu, Angew. Chem. Int. Ed. 57 (2018) 4747-4751. DOI:10.1002/anie.201800767 |
| [17] |
H.J. Zhang, Q. Gu, S.L. You, Org. Lett. 22 (2020) 3297-3301. DOI:10.1021/acs.orglett.0c01109 |
| [18] |
S. Rousseaux, J. García-Fortane, M.A.D.A. Sanchez, S.L. Buchwald, J. Am. Chem. Soc. 133 (2011) 9282-9285. DOI:10.1021/ja203644q |
| [19] |
R.Q. Xu, Q. Gu, W.T. Wu, Z.A. Zhao, S.L. You, J. Am. Chem. Soc. 136 (2014) 15469-15472. DOI:10.1021/ja508645j |
| [20] |
K. Du, P. Guo, Y. Chen, et al., Angew. Chem. Int. Ed. 54 (2015) 3033-3037. DOI:10.1002/anie.201411817 |
| [21] |
R.Q. Xu, P. Yang, C. Zheng, S.L. You, Chin. J. Chem. 38 (2020) 683-689. DOI:10.1002/cjoc.202000109 |
| [22] |
X. Mu, H. Yu, H. Peng, et al., Angew. Chem. Int. Ed. 59 (2020) 8143-8147. DOI:10.1002/anie.202000953 |
| [23] |
J. Nan, Z. Zuo, L. Luo, et al., J. Am. Chem. Soc. 135 (2013) 17306-17309. DOI:10.1021/ja410060e |
| [24] |
L. Yang, H. Zheng, L. Luo, et al., J. Am. Chem. Soc. 137 (2015) 4876-4879. DOI:10.1021/jacs.5b01285 |
| [25] |
D. Yang, L. Wang, M. Kai, et al., Angew. Chem. Int. Ed. 54 (2015) 9523-9527. DOI:10.1002/anie.201503056 |
| [26] |
R.J. Phipps, F.D. Toste, J. Am. Chem. Soc. 135 (2013) 1268-1271. DOI:10.1021/ja311798q |
| [27] |
Q. Yin, S.G. Wang, X.W. Liang, et al., Chem. Sci. 6 (2015) 4179-4183. DOI:10.1039/C5SC00494B |
| [28] |
P. Wang, J. Wang, L. Wang, et al., Adv. Synth. Cata. 360 (2018) 401-405. DOI:10.1002/adsc.201700745 |
| [29] |
H. Egami, R. Rouno, T. Niwa, et al., Angew. Chem. Int. Ed. 59 (2020) 14101-14105. DOI:10.1002/anie.202005367 |
| [30] |
S.G. Wang, Q. Yin, C.X. Zhuo, S.L. You, Angew. Chem. Int. Ed. 54 (2015) 647-650. |
| [31] |
J. Nan, J. Liu, H. Zheng, et al., Angew. Chem. Int. Ed. 54 (2015) 2356-2360. DOI:10.1002/anie.201409565 |
| [32] |
L. Bai, J. Liu, W. Hu, et al., Angew. Chem. Int. Ed. 57 (2018) 5151-5155. DOI:10.1002/anie.201801894 |
| [33] |
X. Ma, J.J. Farndon, T.A. Young, N. Fey, J.F. Bower, Angew. Chem. Int. Ed. 56 (2017) 14531-14535. DOI:10.1002/anie.201708176 |
| [34] |
Y. Zhang, Y. Liao, X. Liu, et al., Chem. Sci. 8 (2017) 6645-6649. DOI:10.1039/C7SC02809A |
| [35] |
P. Zhang, M. Li, X.S. Xue, et al., J. Org. Chem. 81 (2016) 7486-7509. DOI:10.1021/acs.joc.6b01178 |
| [36] |
G.J. Fox, G. Hallas, J.D. Hepworth, K.N. Paskins, Org. Synth. 6 (1988) 181-185. |
| [37] |
J.M. Brittain, P.B.D. de Ia Mare, P.A. Newman, J. Chem. Soc. Perkin Trans. 2 (1981) 32-41. |
| [38] |
D.J. Calvert, P.B.D. de la Mare, H. Suzuki, J. Chem. Soc. Perkin Trans. 2 (1983) 255-260. |
| [39] |
S. Stavber, M. Zupan, J. Org. Chem. 50 (1985) 3609-3612. DOI:10.1021/jo00219a032 |
| [40] |
W.R. Roush, R.J. Neitz, J. Org. Chem. 69 (2004) 4906-4912. DOI:10.1021/jo049426c |
| [41] |
O.A. Attanasi, S. Berretta, C. Favi, et al., Org. Lett. 8 (2006) 4291-4293. DOI:10.1021/ol061637b |
| [42] |
M.P. Hartshorn, H.T. Ing, K.E. Richards, K.H. Sutton, J. Vaughan, Aust. J. Chem. 35 (1982) 1635-1644. DOI:10.1071/CH9821635 |
| [43] |
R.R. Soelch, G.W. Mauer, D.M. Lemal, J. Org. Chem. 50 (1985) 5845-5852. DOI:10.1021/jo00350a079 |
| [44] |
R.G. Clewley, G.G. Cross, A. Fischer, G.N. Henderson, Tetrahedron 45 (1989) 1299-1310. DOI:10.1016/0040-4020(89)80128-0 |
| [45] |
I. Izzo, M. Scioscia, P.D. Gaudio, F.D. Riccardis, Tetrahedron Lett. 42 (2001) 5421-5424. DOI:10.1016/S0040-4039(01)01048-6 |
| [46] |
X. Song, A. Song, F. Zhang, H.X. Li, W. Wang, Nat. Commun. 2 (2011) 524. DOI:10.1038/ncomms1541 |
| [47] |
X. Song, F. Song, X. Meng, P. Ji, W. Wang, Synth Green, Catal. 2 (2021) 377-380. |
| [48] |
L. Wang, D. Yang, D. Li, et al., Chem. Eur. J. 22 (2016) 8483-8487. DOI:10.1002/chem.201601399 |
| [49] |
Q. Yin, S.G. Wang, X.W. Liang, et al., Chem. Sci. 6 (2015) 4179-4183. DOI:10.1039/C5SC00494B |
| [50] |
P. Wang, J. Wang, L. Wang, et al., Adv. Synth. Catal. 360 (2018) 401-405. DOI:10.1002/adsc.201700745 |