 2022, Vol. 33
2022, Vol. 33 


b Henan Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou 450006, China
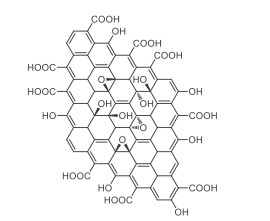
In the past decades, the application of eco-friendly carbonaceous materials as catalysts for organic reactions has gained intensive attention. For example, materials like carbon dots [1, 2], graphitic carbon nitride (g-C3N4) [3-6], have emerged as potent heterogeneous catalysts for various organic transformations. As shown in Scheme 1, graphene oxide (GO) possesses unique structures, various surface functional groups, and excellent properties, such as a large surface area in single or few layers, acidic properties due to carboxyl groups on the edges, oxidative properties assisted by hydroxyl or epoxy on the basal plane [7, 8], conduction band (CB) energy (ECB = 0.51 eV vs. NHE) and valence band (VB) energy (EVB = 3.28–3.98 eV vs. NHE) [9]. Those properties make GO or modified GO as promising materials in biology, energy, organic synthesis, and other fields, like nanocarriers for efficiently impairing tumor mitochondria, heterogeneous carbocatalyst for biomass conversion by controllable depolymerization of lignin [10-13]. Meanwhile, since the significant work achieved by Bielawski in 2010 [14], considerable attention has been paid to organic reactions catalyzed by GO [15-23]. Although multifarious outstanding reviews [24-29] about the synthesis and characterization of GO and modified GO were published, the overview of native GO-catalyzed organic reactions has not been well documented [30-32]. Recently, Bandini et al. summarized the mechanist aspects of organic transformations catalyzed by GO [33]. Considering the importance of GO catalysis, we herein present a minireview aiming to emphasize its significance and widespread applications of GO in organic synthesis over the past decade (mainly from 2011 to 2020). This review includes the application of native graphene oxide for (ⅰ) oxidative reactions, including oxydehydrogenation and oxidative coupling reactions, (ⅱ) functional group transformations, (ⅲ) Friedel-Crafts reactions, (ⅳ) condensation reactions, and so on.

|
Download:
|
| 1. Structure of graphene oxide. | |
{kind=link}
2. Oxidative reactions
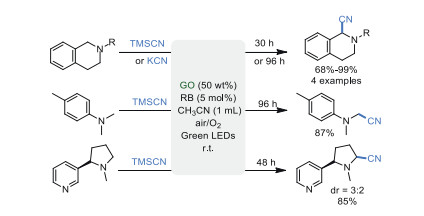
Oxidative reaction, especially oxydehydrogenation, is one of the most common transformations in organic synthesis. The dehydrogenation of N-heterocycles usually requires transition metal and a stoichiometric amount of oxidants [34-37]. In 2017, Gong et al. [38] described an eco-friendly method for the oxydehydrogenation of N-heterocycles catalyzed by metal-free GO in 120 ℃ using air as an oxidant (Scheme 2). This metal-free strategy could be realized through a simple operation. Various valuable compounds, such as 3, 4-dihydroisoquinoline, quinoline, quinazoline, and indole derivatives were successfully synthesized through the activation of O2. Although the reaction needed a high temperature (120 ℃) and long reaction time (up to 96 h in some cases) to achieve an acceptable yield, this work preliminarily demonstrated the roles of various oxygen-containing groups like hydroxyl and epoxy, and π-conjugation on the basal plane of GO.

|
Download:
|
| Scheme 2. Dehydrogenation of nitrogen heterocycles. | |
{kind=link}
Oxidative coupling reaction is one of the most straightforward and efficient strategies for the construction of chemical bonds [39-43]. In 2011, Tan's group [44] demonstrated the potential of GO in visible-light-promoted α-functionalization of tertiary amines. The α-cyanation of tertiary amines was realized by combining high loading of GO and rose bengal (RB) in the presence of visible light (Scheme 3). A series of α-cyanated tertiary amines including N-aryl-tetrahydroisoquinoline, 4-methyl-N, N-dimethylaniline, (S)-nicotine were successfully obtained in good to excellent yields under mild conditions using air as terminal oxidant, avoiding the use of metal catalysts. Meanwhile, it was found that the electron-donating N-protecting groups had a positive influence on reaction rate and the yield of products. Chemoselectivity could be improved significantly, compared with the conditions of RB as photocatalyst alone. Noteworthy, this is the first pioneering work of using GO as a co-catalyst to facilitate the synthesis of small molecular organic compounds under visible light irradiation albeit the role of GO in this system is not clear.

|
Download:
|
| Scheme 3. α-Cyanation of tertiary amines. | |
{kind=link}
In the above-mentioned examples, high loading of GO was generally required to give a good performance. To enhance the catalytic reactivity, Loh and co-workers [45] established a procedure for the preparation of porous graphene oxide (p-GO), in which the as-synthesized GO was treated by base to produce porosity and sequentially acidified to regenerate the water-soluble functional groups. Using p-GO as a catalyst, various transformations of imine intermediates, in situ generated from primary amines, were realized to synthesize α-aminophosphonates, α-aminonitriles, amides, potential bioactive polycyclic heterocompounds, etc., under neat or water conditions (Scheme 4a). Noteworthy, epiminodibenzo[b, f]-[1, 5]diazocine derivatives, an analogous scaffold of Tröger's base, could be synthesized in yield of 75% via a tandem reaction (Scheme 4b).

|
Download:
|
| Scheme 4. Functionalization of primary amines | |
{kind=link}
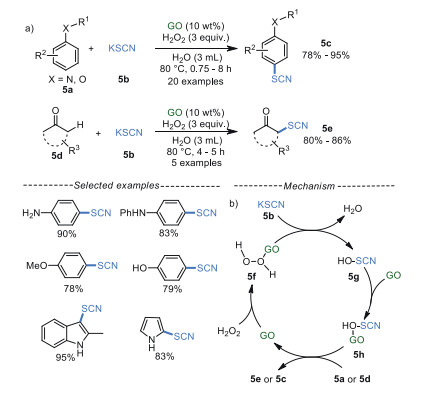
Thiocyanates play pivotal roles in bioactive medicines, materials, pesticides [46], so multiple efforts have been paid in recent years [47-49]. In 2016, Khalili's group [50] developed an environmentally friendly GO-catalyzed regioselective thiocyanation of aromatic amines, phenols, anisoles and carbonyl compounds with KSCN using H2O2 as a green oxidant in water (Schemes 5a). For the indole and pyrrole substrates, the 3-thiocyanated indole and 2-thiocyanated pyrrole were obtained in good to excellent yields. Moreover, the GO catalyst can be reused for five times without significant loss in catalytic activity under the optimized conditions. The plausible mechanism showed that GO surface activate H2O2 to afford 5f, which reacted with KSCN 5b to generate 5g [51, 52], which was reactivated on subsequent reaction with GO to obtain 5h, producing thiocyanium ion SCN+. Eventually, this electrophilic intermediate was attacked by arene 5a or enolizable ketone 5d to produce the target products 5c or 5e (Scheme 5b).

|
Download:
|
| Scheme 5. Thiocyanation of aromatic amines, phenols, anisole and ketones. | |
{kind=link}
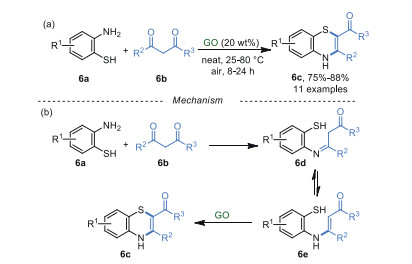
In 2017, Basu et al. [53] synthesized 1, 4-benzothiazines 6c from 2-aminobenzenethiols 6a and 1, 3-diketones 6b using GO as a catalyst in a solvent-free system (Scheme 6a). For the plausible mechanism of this reaction, the condensation between amino 6a and ketones 6b occurred to form an imine 6d and its tautomer 6e. The intermediate 6e was then converted into 1, 4-benzothiazine 6c via oxidative cyclization in the presence of GO and air (Scheme 6b). Unfortunately, substrates 6b with poor electrophilicity and big steric hindrance which are difficult to form the imine intermediates 6d, are not applicable in this procedure. It was known that thiols could be converted into disulfide via oxidative dimerization catalyzed by GO [54]. However, no disulfide was detected in this system.

|
Download:
|
| Scheme 6. Synthesis of functionalized 1, 4-benzothiazines. | |
{kind=link}
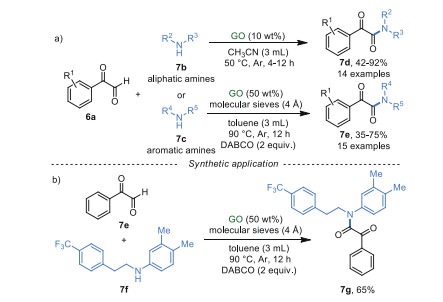
In 2017, Sarma's group [55] described a heterogeneous and metal-free pathway to produce the α-ketoamides and their derivatives from α-ketoaldehyde 7a and aliphatic/aromatic primary and secondary amines in low loading of GO (Scheme 7). This cross-dehydrogenative coupling (CDC) reaction possessed a large substrate scope and the desired products were obtained in moderate to excellent yields. From the control experiments, it was reasoned that GO not only acts as an acidic catalyst but also an oxidative catalyst in this reaction. Additionally, orexin receptor antagonist 7g could be synthesized from phenylglyoxal monohydrate 7e and compound 7f in 65% yield under the optimized conditions (Scheme 7b).

|
Download:
|
| Scheme 7. Synthesis of α-ketoamides and their derivatives. | |
{kind=link}
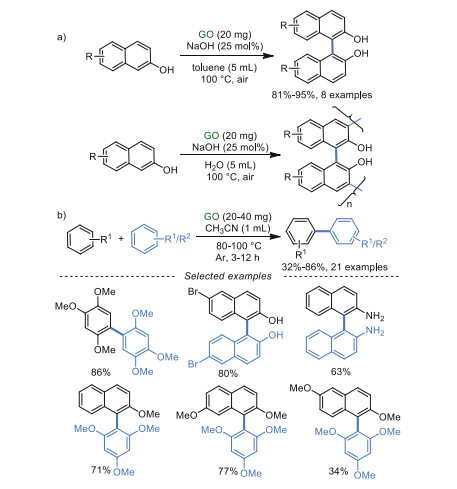
2, 2′-Dihydroxy-1, 1′-binaphthyl is an important compound, which has been widely used as a ligand in asymmetric reactions [56, 57]. Ranganath et al. [58] disclosed that 2, 2′-dihydroxy-1, 1′-binaphthyl can be obtained through the oxidative coupling of 2-naphthols in excellent yields under a low loading of GO (Scheme 8a). Interestingly, solvents can directly control the products. For instance, the homocoupling of 2-naphthols was produced in toluene, while the polymerization of 2-naphthols came out in an aqueous solution. Importantly, NaOH was required as a base in both systems. In 2018, similar work was also reported by Gong et al. [59], which realized the homo-coupling and cross-coupling of aromatics in CH3CN without any additives (Scheme 8b).

|
Download:
|
| Scheme 8. Synthesis of diaryl compounds. | |
{kind=link}
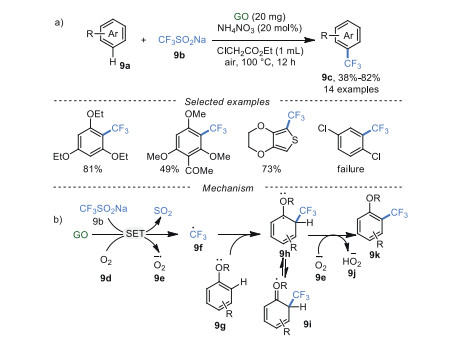
In recent years, a myriad of synthetic methods have been developed for the synthesis of F-containing compounds [60-63], which showed good superiority in lipophilicity, bioavailability and metabolic stability [64-66]. For example, in 2017 Gong's group [67] realized trifluoromethylation of arenes 9a with CF3SO2Na 9b catalyzed by GO in ClCH2CO2Et at 100 ℃ (Scheme 9a). Regretfully, this reaction largely relied on the electronic effect of substrates. Arenes 9a with electron-rich substituents could afford trifluoromethylated arenes 9c in excellent yields, while moderate yields or even traces were observed in the arenes 9a with electronic-poor substituents. To gain deep insight into this reaction, (2, 2, 6, 6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO), a widely used radical scavenger, was added into this reaction under the optimized conditions, only trace targeted product was obtained, indicating that the reaction probably experienced a free radical process. Firstly, unpaired electrons in GO shell can become a radical initiator to generate radical 9f and superoxide radical 9e via tandem single electron transfer (SET) [68]. Subsequently, intermediate 9h was obtained by radical addition of trifluoromethyl radical 9f and arene 9g. Finally, an aromatization reaction was completed to obtain products 9k via hydrogen atom transfer (HAT) of intermediate 9h under the effect of superoxide radical 9e. Additionally, the isomerized intermediate 9i can stabilize the intermediate 9h, and thus facilitated this reaction, which was in agreement with the good reactivity of the substrates with good electron-donating groups.

|
Download:
|
| Scheme 9. Trifluoromethylation of arenes. | |
{kind=link}
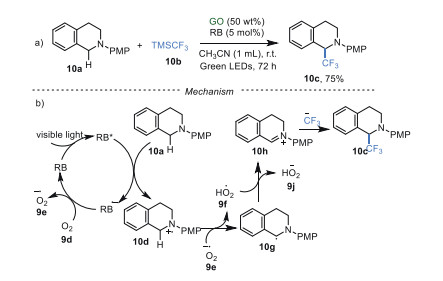
In 2011, Tan's group [44] described a method for the direct α-trifluoromethylation of tertiary amines 10a combining GO and RB as photocatalysts under green LEDs for the first time, utilizing TMSCF3 10b as a CF3 source (Scheme 10a). A plausible pathway showed that RB was transformed into the excited state RB* under the irradiation of visible light. The RB* interacted with the reactant 10a via SET to give the intermediate 10d, along with the generation of radical anion RB•–. Subsequently, RB•– was re-oxidized by oxygen to recover the catalyst RB, accomplishing the photoredox cycle. On the other hand, the intermediate 10d was oxidized by the superoxide radical 9e to give the highly reactive iminium intermediate 10h. Finally, nucleophilic addition of 10h and CF3 anion produce the target compound 10c (Scheme 10b). Although the role of GO was not clear in this process, the control experiment without RB has ruled out the significant oxidability of GO. Additionally, strong π-π interaction between RB and GO did not exist due to electrostatic repulsion, demonstrated by the UV–vis absorption and fluorescence quenching experiments. It was reasoned that the intrinsic acidity and high surface of GO may benefit for this transformation by stabilizing iminium intermediate 10h. Although this work showed the great potential of GO in photocatalysis, high loadings, long reaction time (up to 72 h), and additives were required.

|
Download:
|
| Scheme 10. Trifluoromethylation of 1, 2, 3, 4-tetrahydroisoquinoline derivatives. | |
{kind=link}
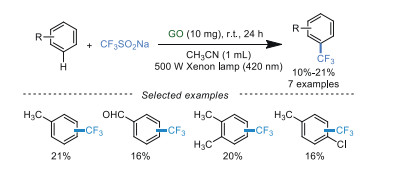
In 2019, Yuan's group [69] reported the visible-light-induced trifluoromethylation of arenes at room temperature using low loading of GO as a catalyst. This protocol avoided additives, metal catalysts, and external photosensitizers (Scheme 11). The results confirmed that GO had great potential in photocatalytic reactions. However, more efforts should be paid to improve the efficiency of photocatalytic reactions.

|
Download:
|
| Scheme 11. Trifluoromethylation of arenes. | |
{kind=link}
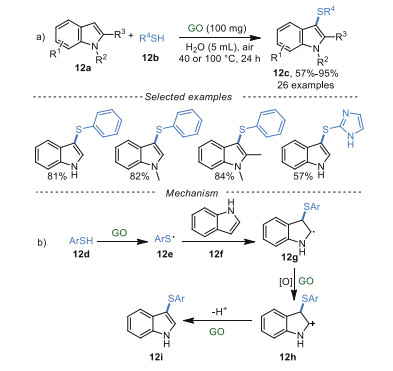
3-Sulfenylindoles exhibit excellent pharmacological properties, including anti-HIV, anti-nociceptive, and anti-allergic activities [70]. In 2019, Wang and Wu's group [71] developed an eco-friendly and atom-economical method to synthesize 3-sulfenylindoles 12c in good to excellent yields via the thiolation of indoles 12a in water (Scheme 12a). The reaction was catalyzed by recyclable GO (up to 10 times) and showed good functional groups tolerance. In particular, N-free indoles were also compatible with this reaction. Control experiments indicated that carboxyl groups (-COOH) and oxygenated groups on GO played vital roles in this reaction. It was proposed that GO could serve as a radical initiator through the functional group on the surface, affording phenylthiophenol radical 12e. Next, radical 12e interacted with 12f to produce the intermediate 12g, which then was oxidized to the intermediate 12h. Finally, the deprotonation of intermediate 12h generated 3-sulfenylindoles 12i as products (Scheme 12b).

|
Download:
|
| Scheme 12. Thiolation of indoles. | |
{kind=link}
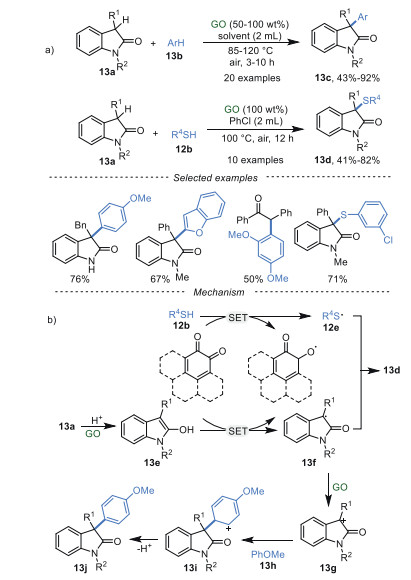
Recently, GO-mediated cross-dehydrogenative coupling of oxindoles 13a with arenes 13b and thiophenols 12b were realized by Su et al. [72] to deliver 3-arylated oxindoles 13c and 3-sulfenylated oxindoles 13d, respectively (Scheme 13a). Notably, N-free oxindoles and deoxybenzoins were also compatible. Meanwhile, the reaction showed good performance on a gram scale. A series of control experiments have been conducted to investigate the reaction pathway. The results indicated that carboxyl groups (-COOH) and oxygenated groups, and quinone-type functionalities in GO played synergistic roles in this transformation. A plausible mechanism was proposed in Scheme 13b. Firstly, the oxindole substrate 13a tautomerized to its enol form 13e due to the intrinsic acidity of GO. The intermediate 13e was rapidly oxidized to the corresponding radical 13f via SET. Then, 13f could undergo a radical-induced coupling with thioyl radical 13e to afford 3-sulfenylated oxindoles 13d. On the other hand, radical 12f was oxidized to the cation 13g catalyzed by GO. Finally, the coupling of 13g and anisole 13h gave the intermediate 13i, followed by deprotonation to generate 3-aryloxindoles 13j.

|
Download:
|
| Scheme 13. Thiolation and arylation of oxindoles. | |
{kind=link}
In 2020, Bhanage's group [73] described a reaction of alcohol 14a and biguanide 14b in the presence of GO and KOH (1 equiv.) at 110 ℃, affording triazines derivatives 14c in moderate to excellent yields (Scheme 14). Simultaneously, GO can be recycled for up to six times without significant loss of catalytic activity (from 91% to 79%). A plausible mechanism of this reaction showed that GO could oxidize alcohols 14a to give the corresponding aldehyde 14d with the aid of a strong base, generating reduced graphene oxide (r-GO), which was re-oxidized by O2. Then aldehyde 14d reacted with biguanide 14b to afford corresponding dihydro-triazine 14e. Finally, the desired product 14c was formed via dehydrogenative aromatization of compound 14e by GO, along with the release of hydrogen.

|
Download:
|
| Scheme 14. Synthesis of triazines and substituted triazines. | |
{kind=link}
3. Functional group transformations 3.1. Oxidation of hydroxyl group
2, 5-Diformylfuran (DFF) can be used as an important building block for the synthesis of furan-containing polymers and materials. It can also be used as a starting material for the synthesis of various poly-Schiff bases, pharmaceuticals, antifungal agents, organic conductors, and cross-linking agents of poly(vinyl alcohol) for battery separations [74]. In recent years, many eco-friendly and renewable pathways for the synthesis of DFF have been reported [75, 76]. In 2015, Hou's group reported a GO-catalyzed transformation of 5-hydroxymethylfurfural (HMF) to DFF. In this reaction, stoichiometric TEMPO was used as an additive in the presence of GO and CH3CN at 100 ℃ (Scheme 15a). Control experiments suggested that the synergistic effect of carboxyl groups (-COOH) and unpaired electrons at GO is responsible for the high selectivity and conversion of this reaction. Following that, the same group [77] described an eco-friendly and metal-free procedure to realize the direct oxidation of fructose into DFF promoted by GO in high temperature in DMSO (Scheme 15b). It was noteworthy that no additive was required in the two steps (dehydration from fructose to HMF; oxidation from HMF to DFF) of this one-pot reaction.

|
Download:
|
| Scheme 15. Selective oxidation of hydroxyl group. | |
{kind=link}
3.2. Oxidation of boric acids
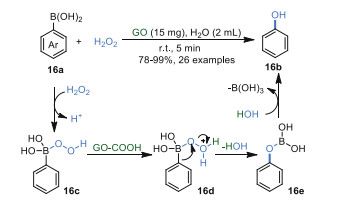
In 2019, Suresh's group [78] realized the ipso-hydroxylation of arylboronic 16a using aqueous H2O2 as a green oxidant and GO as a benign solid-acid catalyst in 5 min (Scheme 16). In this work, GO can be recycled up to five times with a negligible loss of activity. FT-IR, powder X-ray diffraction (PXRD), and transmission electron microscopy (TEM) demonstrated that the structure of GO was maintained. Additionally, the products were obtained in good to excellent yields without time-consuming separation techniques such as chromatography.

|
Download:
|
| Scheme 16. Hydroxylation of arylboronic acids. | |
{kind=link}
From the results of control experiments, a radical mechanism was ruled out and carboxyl groups (-COOH) on the edges of GO were attributed to this reaction. Firstly, electrophilic hydrogen peroxide attacked the boron atom of arylboronic acid 16a, affording intermediate 16c, which was protonated by carboxyl groups (-COOH) on GO to obtain intermediate 16d. Then, consecutive migration of the aryl group from boron to oxygen generated boronate ester 16e. Finally, 16e was converted into the corresponding phenol 16b through hydrolysis.
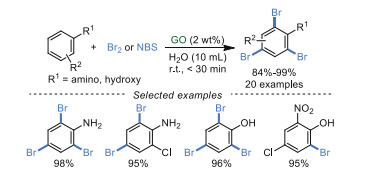
3.3. Oxidative halogenationIn 2018, Shankarling et al. [79] reported the bromination of anilines and phenols using molecular bromine in water catalyzed by GO at room temperature (Scheme 17). These transformations possessed high selectivity for the tri-bromoanilines and tri-bromophenols. Moreover, when N-bromosuccinimide (NBS) as a brominating reagent, GO in this case displayed excellent recyclability without any loss in catalytic activity after several cycles. However, all results of FT-IR, XRD, XPS and elemental analysis demonstrated that GO was reduced to r-GO after the second recycle (the third run) when Br2 was applied as a brominating reagent.

|
Download:
|
| Scheme 17. Bromination of anilines and phenols. | |
{kind=link}
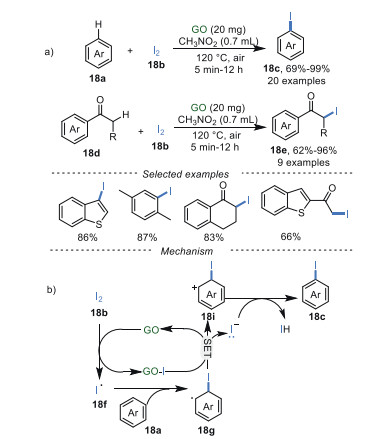
In 2018, Gong's group [80] described GO-catalyzed iodination of arenes 18a and ketones 18d with iodine in CH3NO2 as solvent at 110 ℃ (Scheme 18a). This iodination protocol not only obtained mono-iodination of arenes and ketones with good to excellent yields, regardless of the electronic effects, but also achieved di-iodination of arenes and ketones (10 examples) with moderate to good yields by increasing the amounts of iodine to 4 equiv. When radical scavengers like TEMPO and BHT were added under the standard conditions, the target reaction was suppressed suggesting a radical pathway. A plausible mechanism showed that the unpaired electrons on GO surface played significant roles in the initiation of iodine radical 18f. Then the addition of 18f to aromatic ring 18a obtains the aromatic radical 18g, which undergoes a SET process to obtain aromatic cation 18i, along with the recovery of GO catalyst. Afterward, the desired products 18c were produced through deprotonation and aromatization (Scheme 18b). Unfortunately, GO could not be reused in this reaction.

|
Download:
|
| Scheme 18. Iodination of arenes and ketones. | |
{kind=link}
4. Friedel-Crafts reactions
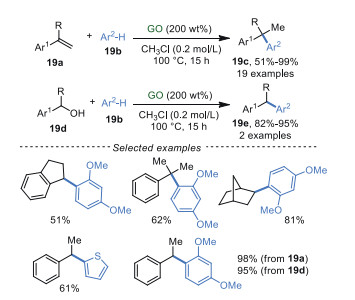
In 2015, He and Szostak et al. [81] developed an atom economic reaction of arenes 19b and styrene 19a or arylmethanols 19d to obtain diarylalkanes 19c and 19e (Scheme 19). This protocol promoted by GO was carried out under metal-free and additive-free conditions with good functional group tolerance and excellent regioselectivity to deliver the corresponding products in high yields (up to 99%), however, high loading of GO (up to 200 wt%) was still needed in this reaction.

|
Download:
|
| Scheme 19. Alkylation of arenes with styrenes or alcohols. | |
{kind=link}
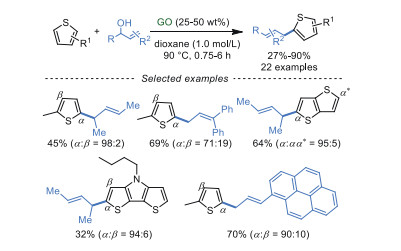
Similarly, the allylic alkylation of thiophenes with alcohols was realized by Bandini and co-workers [82] under the assistance of GO catalyst (Scheme 20). This procedure can tolerate myriad functional groups in dioxane as solvent at 90 ℃, generating the corresponding products in moderate to excellent yields (up to 90%). The control experiments indicated that GO was significant for this reaction, owing to the cooperative effect of the π-conjugated system and functional groups (carboxyl groups, epoxide moieties) on GO.

|
Download:
|
| Scheme 20. Allylic alkylation of thiophenes with alcohols. | |
{kind=link}
Moreover, GO also exhibited efficient catalytic activity for the synthesis of 3, 3′-bisindolylmethane derivatives from indole derivatives 21a and ethers at room temperature and air atmosphere (Scheme 21a) [83]. The cyclic and linear benzyl ethers, including tetrahydrofuran (THF), 1, 3-dihydroisobenzofuran, isochromane, (methoxymethyl)benzene, and diethyl ether, proved to be suitable in this procedure giving remarkable regioselectivities and moderate to excellent yields. The reaction pathway was proposed as shown in Scheme 21b. Firstly, THF was oxidized into intermediate 21d in the presence of GO and oxygen, which was further dehydrated to obtain intermediate 21e due to the intrinsic acidity of GO. Then, 21e was protonated to give the intermediate 21f, which reacted with indole 12f to generate intermediate 21h. Subsequently, 21h was converted into 21i, which reacted with indole 12f to give the target product 21j via the Friedel-Crafts alkylation.

|
Download:
|
| Scheme 21. Double indolation of ethers. | |
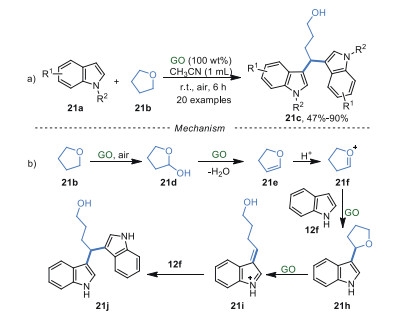{kind=link}
5. Condensation reactions
In 2014, Sarma et al. [84] designed a simple, efficient, methodology to synthesize α-aminophosphonates and 3, 4-dihydropyrimidin-2-ones via a three-component one-pot reaction, catalyzed by low loading of GO in a solvent-free environment under ultrasonication (Scheme 22). This pathway demonstrated for the first time that GO can be used as a mild, nontoxic, and sustainable catalyst for the multicomponent coupling reaction. Through a series of control experiments, it could be concluded that the formation of the imine intermediate was the critical step. Although GO catalyst displayed excellent catalytic activity after 7 runs, it was demonstrated that the structure of recycled GO was damaged from FT-IR, UV-visible studies, and thermal gravimetric analysis (TGA). Simultaneously, oxygen played a negligible role in this coupling, and GO nanosheets acted as a facilitator to stabilize the imine intermediate.

|
Download:
|
| Scheme 22. Synthesis of α-aminophosphonates and 3, 4-dihydropyrimidin-2-ones. | |
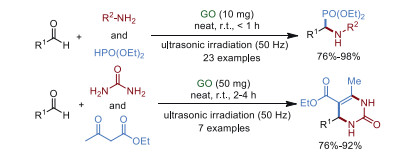{kind=link}
The GO-mediated direct 2-alkylation of 1, 3-dicarbonyl compounds 23a was developed by He and Szostak et al. [85] with high reactivity, excellent regioselectivity using abundant olefins 23b and alcohols 23d as alkylating agents (Scheme 23a). Additionally, the results of FT-IR, XPS indicated that polar functional groups had negligible changes on the GO. The proposed mechanism was shown in Scheme 23b. The activation of both coupling partners 23a and 23f via transient coordination with GO surface was the critical step. Then, due to the presence of polar in intermediate 23k, target compound 23l was produced by releasing water and catalyst GO.

|
Download:
|
| Scheme 23. 2-Alkylation of 1, 3-dicarbonyl compounds. | |
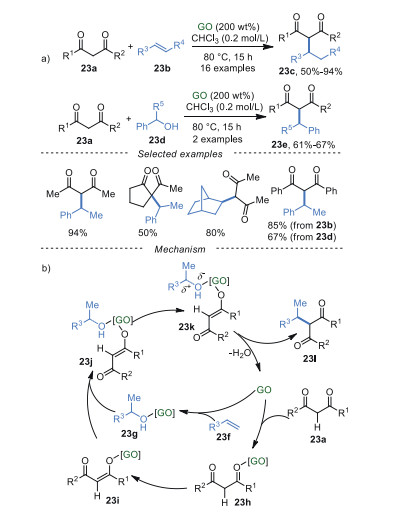{kind=link}
6. Other reactions
In 2016, Wang and Ma's group [86] developed a heterogeneous and direct C-H arylation of aryl iodides in low loading of GO at 120 ℃ to give biaryl compounds as products (Scheme 24). The additive KOt-Bu was significant for the transformation because K+ ions can be stabilized and activated by negatively charged oxygen atoms, facilitating the activation of the C-I bond. Generally, the synergistic effects of K+ ions, oxygen-containing groups on GO, and giant π-conjugated system of GO were greatly contributed to direct C-H arylation of benzene. However, other aryl halides like aryl bromides and aryl chlorides were not suitable in this protocol.

|
Download:
|
| Scheme 24. Synthesis of biaryl compounds. | |
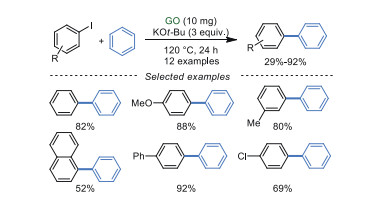{kind=link}
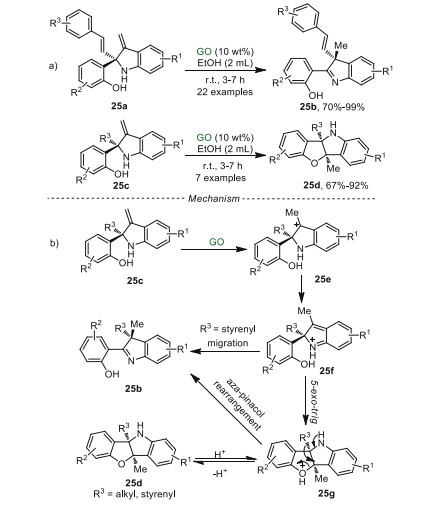
The construction of quaternary carbon center at the C2- and C3-positions of indoline moieties attracted huge attention [87]. Recently, Su and Mo et al. [88] used GO as a heterogeneous catalyst to synthesize quaternary carbon center-containing 2-(indol-2-yl)phenols 25b and benzofuro[3, 2-b]indolines 25d in EtOH at ambient temperature for the first time (Scheme 25a). The proposed reaction pathway was shown in Scheme 25b. Firstly, the electrophilic addition between the acidic group of GO and 25c generated the intermediate 25e. Then, intermediate 25e isomerized to a stable aza-ortho-xylylene intermediate 25f. The results of Raman spectroscopy and PXRD revealed that the intermediates 25e and 25f could be stabilized by hydroxyl and epoxy on GO. When R3 was styrenyl group, intermediate 25f was inclined to undergo a 1, 2-styrenyl migration to generate product 25b, releasing a proton to complete this catalytic cycle. Alternatively, intermediate 25f could undergo 5-exo-trig cyclization to obtain intermediate 25g, which released a proton to produce compound 25d Interestingly, when R3 was styrenyl group, compound 25d can be reactivated by proton to regenerate intermediate 25g, which could be further transformed into compound 25b through an aza-pinacol rearrangement. It was noteworthy that the stereochemistry of the C3-quaternary carbon center generated from the C2-quaternary carbon center of indoline was retained. Compared with the previous reports [89, 90], the GO catalyst of this heterogeneous system could be reutilized at least seven times without obvious loss in catalytic activity.

|
Download:
|
| Scheme 25. Construction of stereochemical indoline moieties. | |
{kind=link}
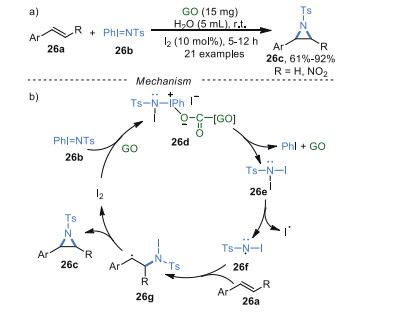
In 2017, Rai and co-workers [91] reported a GO/I2-promoted aziridination of olefins in water. This nitrene insertion reaction provided tosylaziridines as products at room temperature (Scheme 26a). A plausible mechanism was given in Scheme 26b. Initially, PhI=NTs 26b reacted with I2 with the catalysis of GO to generate the key intermediate N, N-diiodotosylamide 26e. Subsequently, the intermediate 26e underwent a homolytic cleavage of N-I bond to produce amidyl radical 26f. Then, radical 26f added to the olefin 26a to generate intermediate 26g, which underwent intramolecular cyclization to produce target molecule 26c, along with the regeneration of I2 for the next catalytic cycle. The GO catalyst could be reused up to six times.

|
Download:
|
| Scheme 26. Construction of tosylaziridines. | |
{kind=link}
7. Conclusion
Graphene oxide (GO) as a kind of inexpensive carbon material can be employed as a metal-free carbocatalyst and efficient acidic catalyst for various organic reactions. In this minireview, we summarized the recent advances (mainly from 2011 to 2020) of native GO-promoted organic reactions including oxidative coupling reactions, functional group transformations, oxidative halogenation, condensation reactions, and so on. Notably, in those reported systems GO could be recycled and reused for several times. However, the employment of GO as a light absorber and heterogeneous photocatalyst in visible-light-induced organic reactions is still rare. We believe that the application of graphene oxide as a heterogeneous photocatalyst in photocatalytic organic transformations will gain more attention in the coming future.
Declaration of competing interestThe authors declare no conflict of interest.
AcknowledgmentsWe acknowledge the financial support from the National Natural Science Foundation of China (Nos. 21971224, 22171249), and the Natural Science Foundation of Henan Province (No. 202300410375).
| [1] |
W.D. Li, Y. Liu, B.Y. Wang, et al., Chin. Chem. Lett. 30 (2019) 2323-2327. DOI:10.1016/j.cclet.2019.06.040 |
| [2] |
S.J. Phang, L.L. Tan, Catal. Sci. Technol. 9 (2019) 5882-5905. DOI:10.1039/C9CY01452G |
| [3] |
Y.F. Si, X.L. Chen, X.Y. Fu, et al., ACS Sustain. Chem. Eng. 8 (2020) 10740-10746. |
| [4] |
S.J. Chen, T.F. Niu, B.Q. Ni, Chin. J. Synth. Chem. 29 (2021) 562-569. |
| [5] |
M.H. Muhammad, X.L. Chen, Y. Liu, et al., ACS Sustain. Chem. Eng. 8 (2020) 2682-2687. DOI:10.1021/acssuschemeng.9b06010 |
| [6] |
F.L. Zeng, H.L. Zhu, X.L. Chen, L.B. Qu, B. Yu, Green Chem. 23 (2021) 3677-3682. DOI:10.1039/D1GC00938A |
| [7] |
B.F. Machado, P. Serp, Catal. Sci. Technol. 2 (2012) 54-75. DOI:10.1039/C1CY00361E |
| [8] |
H.L. Wang, H.J. Dai, Chem. Soc. Rev. 42 (2013) 3088-3113. DOI:10.1039/c2cs35307e |
| [9] |
I. Shown, H.C. Hsu, Y.C. Chang, et al., Nano Lett. 14 (2014) 6097-6103. DOI:10.1021/nl503609v |
| [10] |
J.L. Li, H.C. Bao, X.L. Hou, et al., Angew. Chem. Int. Ed. 51 (2012) 1830-1834. DOI:10.1002/anie.201106102 |
| [11] |
H.Q. Zhu, B. Zhang, N.L. Zhu, M.C. Li, Q.L. Yu, Chin. Chem. Lett. 32 (2021) 1220-1223. DOI:10.1016/j.cclet.2020.09.003 |
| [12] |
Y.J. Song, W.L. Wei, X.G. Qu, Adv. Mater. 23 (2011) 4215-4236. DOI:10.1002/adma.201101853 |
| [13] |
J.J. Zeng, Z.H. Tong, H.X. Bao, et al., Fuel 267 (2020) 117100. DOI:10.1016/j.fuel.2020.117100 |
| [14] |
D.R. Dreyer, H.P. Jia, C.W. Bielawski, Angew. Chem. Int. Ed. 49 (2010) 6813-6816. |
| [15] |
M.R. Acocella, M. Mauro, L. Falivene, L. Cavallo, G. Guerra, ACS Catal. 4 (2014) 492-496. DOI:10.1021/cs401053t |
| [16] |
V.D. Ebajo, C.R.L. Santos, G.V. Alea, Y.A. Lin, C.H. Chen, Sci. Rep. 9 (2019) 15579. DOI:10.1038/s41598-019-51833-2 |
| [17] |
W.Z. Zhu, F.R. Tao, S.W. Chen, et al., ACS Sustain. Chem. Eng. 7 (2019) 296-305. DOI:10.1021/acssuschemeng.8b03373 |
| [18] |
L. Lombardi, D. Bellini, A. Bottoni, et al., Chem. Eur. J. 26 (2020) 10427-10432. DOI:10.1002/chem.202001373 |
| [19] |
Y.R. Girish, S. Pandit, S. Pandit, M. De, Chem. Asian J. 12 (2017) 2393-2398. DOI:10.1002/asia.201701072 |
| [20] |
S.M. Islam, A.S. Roy, R.C. Dey, S. Paul, J. Mol. Catal. A: Chem. 394 (2014) 66-73. DOI:10.1016/j.molcata.2014.06.038 |
| [21] |
Y.H. Wang, R. Sang, Y. Zheng, et al., Catal. Commun. 89 (2017) 138-142. DOI:10.1016/j.catcom.2016.09.027 |
| [22] |
D. Khalili, S. Lavian, M. Moayyed, Tetrahedron Lett. 61 (2020) 151470. DOI:10.1016/j.tetlet.2019.151470 |
| [23] |
H.R. Wu, C.L. Su, R. Tandiana, et al., Angew. Chem. Int. Ed. 57 (2018) 10848-10853. DOI:10.1002/anie.201802548 |
| [24] |
D. Chen, H.B. Feng, J.H. Li, Chem. Rev. 112 (2012) 6027-6053. DOI:10.1021/cr300115g |
| [25] |
D.R. Dreyer, A.D. Todd, C.W. Bielawski, Chem. Soc. Rev. 43 (2014) 5288-5301. DOI:10.1039/C4CS00060A |
| [26] |
S. Eigler, A. Hirsch, Angew. Chem. Int. Ed. 53 (2014) 7720-7738. DOI:10.1002/anie.201402780 |
| [27] |
P.P. Brisebois, M. Siaj, J. Mater. Chem. C 8 (2020) 1517-1547. DOI:10.1039/C9TC03251G |
| [28] |
Q.S. Mei, B.H. Liu, G.M. Han, et al., Adv. Sci. 6 (2019) 1900855. DOI:10.1002/advs.201900855 |
| [29] |
H.B. Jie, S. Tao, H.X. Yu, Y. Mao, Q.W. Hao, Chin. J. Org. Chem. 40 (2020) 3279-3288. DOI:10.6023/cjoc202004022 |
| [30] |
O. Mohammadi, M. Golestanzadeh, M. Abdouss, New J. Chem. 41 (2017) 11471-11497. DOI:10.1039/C7NJ02515G |
| [31] |
M.S. Ahmad, Y. Nishina, Nanoscale 12 (2020) 12210-12227. DOI:10.1039/D0NR02984J |
| [32] |
C.L. Su, K.P. Loh, Acc. Chem. Res. 46 (2013) 2275-2285. DOI:10.1021/ar300118v |
| [33] |
L. Lombardi, M. Bandini, Angew. Chem. Int. Ed. 59 (2020) 20767-20778. DOI:10.1002/anie.202006932 |
| [34] |
X.J. Cui, Y.H. Li, S. Bachmann, et al., J. Am. Chem. Soc. 137 (2015) 10652-10658. DOI:10.1021/jacs.5b05674 |
| [35] |
E. Zhang, H.W. Tian, S.D. Xu, X.C. Yu, Q. Xu, Org. Lett. 15 (2013) 2704-2707. DOI:10.1021/ol4010118 |
| [36] |
S. Bera, A. Bera, D. Banerjee, Org. Lett. 22 (2020) 6458-6463. DOI:10.1021/acs.orglett.0c02271 |
| [37] |
N.O. Balayeva, Z. Mamiyev, R. Dillert, N. Zheng, D.W. Bahnemann, ACS Catal. 10 (2020) 5542-5553. DOI:10.1021/acscatal.0c00556 |
| [38] |
J.Y. Zhang, S.Y. Chen, F.F. Chen, et al., Adv. Synth. Catal. 359 (2017) 2358-2363. DOI:10.1002/adsc.201700178 |
| [39] |
K. Sun, F. Xiao, B. Yu, W.M. He, Chin. J. Catal. 42 (2021) 1921-1943. DOI:10.1016/S1872-2067(21)63850-0 |
| [40] |
C.H. Ma, M. Chen, Z.W. Feng, et al., New J. Chem. 45 (2021) 9302-9314. DOI:10.1039/D1NJ00704A |
| [41] |
S.H. Tian, T. Luo, Y.P. Zhu, J.P. Wan, Chin. Chem. Lett. 31 (2020) 3073-3082. DOI:10.1016/j.cclet.2020.07.042 |
| [42] |
G.P. Yang, F. Y.f.Liu, K. Li, et al., Chin. Chem. Lett. 31 (2020) 3233-3236. DOI:10.1016/j.cclet.2020.07.018 |
| [43] |
K.D. Zhou, J.P. Huang, J. Wu, G.Y.S. Qiu, Chin. Chem. Lett. 32 (2021) 37-39. DOI:10.1016/j.cclet.2020.11.049 |
| [44] |
Y.H. Pan, S. Wang, C.W. Kee, et al., Green Chem. 13 (2011) 3341-3344. DOI:10.1039/c1gc15865a |
| [45] |
C.L. Su, R. Tandiana, J. Balapanuru, et al., J. Am. Chem. Soc. 137 (2015) 685-690. DOI:10.1021/ja512470t |
| [46] |
M. Artico, R. Silvestri, E. Pagnozzi, et al., J. Med. Chem. 43 (2000) 1886-1891. DOI:10.1021/jm9901125 |
| [47] |
A. Yadav Qumruddeen, R. Kant, C.B. Tripathi, J. Org. Chem. 85 (2020) 2814-2822. DOI:10.1021/acs.joc.9b03275 |
| [48] |
D. Chitteti, P. Pannala, G.U. Vinod, R. Pedavenkatagari Narayana, Curr. Org. Synth. 18 (2021) 233-247. DOI:10.2174/1570179417999201203211855 |
| [49] |
Y.Y. Shan, L. Su, D.P. Chen, et al., Chin. Chem. Lett. 32 (2021) 437-440. DOI:10.1016/j.cclet.2020.04.041 |
| [50] |
D. Khalili, New J. Chem. 40 (2016) 2547-2553. DOI:10.1039/C5NJ02314A |
| [51] |
J.J. Barnett, D.M. Stanbury, Inorg. Chem. 41 (2002) 164-166. DOI:10.1021/ic015597d |
| [52] |
J.N. Figlar, D.M. Stanbury, Inorg. Chem. 39 (2000) 5089-5094. DOI:10.1021/ic000561r |
| [53] |
S. Bhattacharya, P. Ghosh, B. Basu, Tetrahedron Lett. 58 (2017) 926-931. DOI:10.1016/j.tetlet.2017.01.068 |
| [54] |
D.R. Dreyer, H.P. Jia, A.D. Todd, J.X. Geng, C.W. Bielawski, Org. Biomol. Chem. 9 (2011) 7292-7295. DOI:10.1039/c1ob06102j |
| [55] |
B. Majumdar, D. Sarma, T. Bhattacharya, T.K. Sarma, ACS Sustainable Chem. Eng. 5 (2017) 9286-9294. DOI:10.1021/acssuschemeng.7b02267 |
| [56] |
S.N. Richter, S. Maggi, S.C. Mels, M. Palumbo, M. Freccero, J. Am. Chem. Soc. 126 (2004) 13973-13979. DOI:10.1021/ja047655a |
| [57] |
D. Verga, M. Nadai, F. Doria, et al., J. Am. Chem. Soc. 132 (2010) 14625-14637. DOI:10.1021/ja1063857 |
| [58] |
M. Shaikh, A. Sahu, A. Kiran Kumar, et al., Green Chem. 19 (2017) 4533-4537. DOI:10.1039/C7GC02227A |
| [59] |
J.X. Fang, Z.Y. Peng, Y. Yang, et al., Asian J. Org. Chem. 7 (2018) 355-358. DOI:10.1002/ajoc.201700673 |
| [60] |
Y. Liu, X.L. Chen, K. Sun, et al., Org. Lett. 21 (2019) 4019-4024. DOI:10.1021/acs.orglett.9b01175 |
| [61] |
M.W.W. Michael, P.S. James, W.K. Jeff, K.S. Nathaniel, J. Am. Chem. Soc. 142 (2020) 18698-18705. DOI:10.1021/jacs.0c09505 |
| [62] |
N. Naoki, K. Takashi, A. Munetaka, ACS Catal. 9 (2019) 4382-4387. DOI:10.1021/acscatal.9b00473 |
| [63] |
S.P. Lv, Y.F. Sun, Y. Xu, S.H. Yang, L. Wang, Chin. Chem. Lett. 31 (2020) 1568-1571. DOI:10.1016/j.cclet.2019.11.027 |
| [64] |
J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432-2506. DOI:10.1021/cr4002879 |
| [65] |
K. Haranahalli, T. Honda, I. Ojima, J. Fluorine. Chem. 217 (2019) 29-40. DOI:10.1016/j.jfluchem.2018.11.002 |
| [66] |
H. Mei, A.M. Remete, Y.P. Zou, et al., Chin. Chem. Lett. 31 (2020) 2401-2413. DOI:10.1016/j.cclet.2020.03.050 |
| [67] |
J.Y. Zhang, Y. Yang, J.X. Fang, G.J. Deng, H. Gong, Chem. Asian J. 12 (2017) 2524-2527. DOI:10.1002/asia.201700939 |
| [68] |
C.L. Su, M. Acik, K. Takai, et al., Nat. Commun. 3 (2012) 1298. DOI:10.1038/ncomms2315 |
| [69] |
Y. Tong, H. Pan, W.C. Huang, et al., New J. Chem. 43 (2019) 8741-8745. DOI:10.1039/C9NJ01188A |
| [70] |
C. Shen, P.F. Zhang, Q. Sun, et al., Chem. Soc. Rev. 44 (2015) 291-314. DOI:10.1039/C4CS00239C |
| [71] |
M. Chen, Y. Luo, C. Zhang, et al., Org. Chem. Front. 6 (2019) 116-120. DOI:10.1039/C8QO00726H |
| [72] |
H.R. Wu, C.T. Qiu, Z.F. Zhang, et al., Adv. Synth. Catal. 362 (2020) 789-794. DOI:10.1002/adsc.201901224 |
| [73] |
S.R. Chaurasia, R. Dange, B.M. Bhanage, Catal. Commun. 137 (2020) 105933. DOI:10.1016/j.catcom.2020.105933 |
| [74] |
J.P. Ma, Z.T. Du, J. Xu, Q.H. Chu, Y. Pang, ChemSusChem 4 (2011) 51-54. DOI:10.1002/cssc.201000273 |
| [75] |
M. Zhang, Z. Li, X. Xin, et al., ACS Catal. 10 (2020) 14793-14800. DOI:10.1021/acscatal.0c04330 |
| [76] |
Z.L. Yuan, B. Liu, P. Zhou, Z.H. Zhang, Q. Chi, Catal. Sci. Technol. 8 (2018) 4430-4439. DOI:10.1039/C8CY01246F |
| [77] |
G.Q. Lv, H.L. Wang, Y.X. Yang, et al., Green Chem. 18 (2016) 2302-2307. DOI:10.1039/C5GC02794B |
| [78] |
M. Karthik, P. Suresh, ACS Sustainable Chem. Eng. 7 (2019) 9028-9034. DOI:10.1021/acssuschemeng.9b01361 |
| [79] |
P.V. Ghorpade, D.A. Pethsangave, S. Some, G.S. Shankarling, J. Org. Chem. 83 (2018) 7388-7397. DOI:10.1021/acs.joc.8b00188 |
| [80] |
J.Y. Zhang, S.G. Li, G.J. Deng, H. Gong, ChemCatChem 10 (2018) 376-380. DOI:10.1002/cctc.201701182 |
| [81] |
F. Hu, M. Patel, F. Luo, et al., J. Am. Chem. Soc. 137 (2015) 14473-14480. DOI:10.1021/jacs.5b09636 |
| [82] |
L. Favaretto, J. An, M. Sambo, et al., Org. Lett. 20 (2018) 3705-3709. DOI:10.1021/acs.orglett.8b01531 |
| [83] |
X.J. Peng, Y. Zen, Q. Liu, L.X. Liu, H.S. Wang, Org. Chem. Front. 6 (2019) 3615-3619. DOI:10.1039/C9QO00926D |
| [84] |
T. Bhattacharya, B. Majumdar, D. Dey, T.K. Sarma, RSC Adv. 4 (2014) 45831-45837. DOI:10.1039/C4RA08533G |
| [85] |
G.R. Meng, M. Patel, F.X. Luo, et al., Chem. Commun. 55 (2019) 5379-5382. DOI:10.1039/C9CC02578B |
| [86] |
Y.J. Gao, P. Tang, H. Zhou, et al., Angew. Chem. Int. Ed. 55 (2016) 3124-3128. DOI:10.1002/anie.201510081 |
| [87] |
Y.W. Chen, X. Zhang, X.J. Wang, et al., J. Am. Chem. Soc. 143 (2021) 4302-4310. DOI:10.1021/jacs.0c12963 |
| [88] |
H.P. Zhao, G.C. Liang, S.M. Nie, et al., Green Chem. 22 (2020) 404-410. DOI:10.1039/C9GC03345A |
| [89] |
W.D. Liu, G.Q. Xu, X.Q. Hu, P.F. Xu, Org. Lett. 19 (2017) 6288-6291. DOI:10.1021/acs.orglett.7b02989 |
| [90] |
Y.Y. Yu, J.W. Li, L. Jiang, J.R. Zhang, L.S. Zu, Angew. Chem. Int. Ed. 56 (2017) 9217-9221. DOI:10.1002/anie.201705539 |
| [91] |
P. Shukla, S. Mahata, A. Sahu, et al., RSC Adv. 7 (2017) 48723-48729. DOI:10.1039/C7RA09351A |