 2022, Vol. 33
2022, Vol. 33 


b College of Medicine, Southwest Jiaotong University, Chengdu 610031, China;
c Emergency Intensive Care Unit, Personalized Drug Therapy Key Laboratory of Sichuan Province, Sichuan Academy of Medical Science & Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610072, China
Protein kinase is the most effective drug target in cancer treatment since the systematic regulation of protein kinase activity is closely related to tumorigenesis [1, 2]. Therefore, targeting protein kinases has always been an important direction for drug discovery [3]. Dual specificity tyrosine phosphorylation regulatory kinase (DYRK), an evolutionarily conserved protein kinase family directed by proline or arginine [4], which together with cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinase (GSK) and CDK-like kinases, are collectively termed the CMGC family. The DYRK family has five members, DYRK1A, DYRK1B, DYRK2, DYRK3 and DYRK4, and all these kinases could regulate cell proliferation, survival and differentiation. DYRK1A and DYRK1B are located in the nucleus, which are the most studied members [5-9]. DYRK1B is expressed at a low level in most tissues except in ovarian and pancreatic cancer [10, 11]. DYRK1A is essential for brain development and function formation [12, 13], whose phosphorylation promotes the development of neuronal dendrites and dendrites by regulating the growth of microfibers and actin assemblies via Tau and N-WASP proteins [14-16]. DYRK1A is located in the exclusive trisomy region of human chromosome 21 (Hsa21) [17, 18], which participates in many biological processes such as neural development and performs key functions by phosphorylating downstream substrates transcription factors, splicing factors and synaptic proteins [19]. DYRK1A is associated with congenital heart disease, Alzheimer's disease (AD) [20], diabetes [21] and other diseases for its important role in brain development and functional regulation [22-26], neurodegenerative diseases, tumorigenesis and apoptosis and other cellular functions [22, 27-30].
2. DYRK1A molecular structure and biological functionThe DYRK1A gene has 151 kb, 15 exons, which encodes two protein subtypes at positions 763 and 754. And DYRK1A has a functional dichotomous nuclear localization signal (NLS) at the N terminus of the DH-box, which is the second NLS between the X and XI subdomains in the kinase domain [18]. DYRK1A autophosphorylates Tyr21 residues in the activation loop to stabilize its active conformation [31]. DYRK1A can phosphorylate excessive protein targets on serine or threonine residues in mature cells, which brings the DYRK1A gene a variety of biological functions [32]. The effect of DYRK1A on cell cycle and differentiation regulation, T cell regulation, cytoscaffold stability, brain nerve development and synaptic activity are disrupted when the expression of DYRK1A increases, which leads to a variety of diseases [31, 33-37]. Therefore, inhibition of DYRK1A is one of the main therapeutics for diseases such as AD, cancer, inflammation and diabetes.
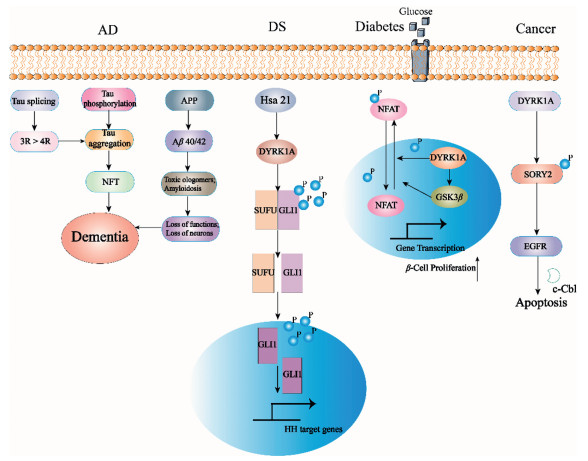
3. DYRK1A and diseases 3.1. ADAD is one of the leading causes of death worldwide due to the aging population, which is a neurodegenerative disease caused by β-amyloid (Aβ) deposition and neurofibrillary nodules [38-40]. And these plaques are neurotoxic peptides produced by insoluble fibers (gathered by Aβ 40 and Aβ 42), β-and γ-secretase hydrolyzing amyloid precursor protein (APP). The level of α secretase is significantly decreased in AD patients, and the accumulation of β secretase components has been observed in animal models and AD patients. DYRK1A is highly expressed in the hippocampus in neurodegenerative diseases, which induces the production of Aβ by altering APP phosphorylation. The above evidence indicates that DYRK1A is a potential target for the treatment of AD [41-44]. DYRK1A phosphorylates Tau (including Thr181, Thr212 and Thr231) in NFT of the brain and APP in Thr668 or presenilin 1 (PS1) in Thr354 in AD patients. DYRK1A also phosphorylates some AD-related immune mediators, including calcineurin nuclear factor (NFAT), which activates T cells, and transcriptional activator-3 (STAT3) that is involved in signal transduction [34, 45-49]. In addition, some studies have shown that inhibition of DYRK1A can restore the balance of 3R/4R Tau protein by blocking exons in Tau mRNA, thus promoting Tau aggregation, which is one of the main markers of AD [50-52]. And the is study shows that DYRK1A inhibitor can improve the pathological changes of the mouse AD model [53]. Harmine, a potent DYRK1A inhibitor, could inhibit DYRK1A-mediated Tau phosphorylation [half maximal inhibitory concentration (IC50) = 0.7 µmol/L)] [54].
3.2. DSDS is a complex cognitive disorder caused by human chromosome 21 trisomy [55, 56]. In the United States, about 1 in 300 babies carry trisomy, half of which are chromosome 21 trisomy that can cause DS. DS not only causes intellectual disability, but also leads to congenital heart disease, hematopoietic disorders and early-onset AD [57-59], which cannot be ignored. But what is interesting is that the incidence of most solid tumors in patients with DS is greatly reduced [60], which is worthy for anti-tumor researches paying attention to. DS is characterized by intellectual disability and significant obstacles to planning, decision-making and memory functions, which may be caused by abnormalities in the hippocampus and medial prefrontal cortex [61-67]. Single gene dose increase in Hsa21 (such as DYRK1A) is thought to explain many neurodevelopmental and abnormal phenotypic changes associated with DS [68-70], which has become a target for the treatment of DS. Hedgehog (Hh) and mTOR, essential signal pathways for the embryonic development and cellular metabolism, both are coordinated by the primary cilium. GLI1, a key downstream transcription effector of the Hh signaling pathway, is involved in cell growth, differentiation and tissue patterning in embryonic development, of which the phosphorylation and nucleus localization is conducted by DYRK1A. And studies have shown that the phosphorylation of Ser408 on GLI1 was blocked with the presence of the selective DYRK1A inhibitor harmine. In addition, the activity of mTORC1 is also negatively regulated by DYRK1A. In another word, inhibiting DYRK1A is of positive meaning for the treatment of DS [71-78].
3.3. DiabetesDiabetes is a group of metabolic diseases characterized by hyperglycemia. There are more than 400 million diabetic patients worldwide, which is equivalent to one diabetic patient in every 12 people, and the prevalence is still rising [79]. Among them, 90% of these cases are type 2 diabetes with elevated blood sugar levels caused by insufficient insulin secretion or insulin resistance. Absolute or relative deficiency of insulin secretion due to functional impairment of islet B cells (belong to endocrine cells, about 70% of all islets), which lead to type 1 and type 2 diabetes. Recent research has identified DYRK1A as a drug target in the genomics study of insulinoma, for which can regulate the cell cycle of human β cells and may be a key molecule for the proliferation of β cell [7, 80]. DYRK1A inhibitors can activate the nuclear factor of T cells, thereby allowing the acquisition of promoters that activate human β cell proliferation genes, which could bring therapeutic effect to type 1 and type 2 diabetes. Studies have shown that harmine may activate NFAT of the T cell transcription factor family, which activates the cell cycle mechanism and thus maintains differentiation in human β cells. And this mechanism was also demonstrated in different mouse, rat and human islet implantation models [25]. In addition, harmine can induce the marker index of human β cells in the range of 1%–3%. Therefore, DYRK1A inhibitor is a novel direction for the treatment of diabetes.
3.4. CancerResearchers accidentally found that the incidence of cancer in patients with DS is significantly lower than that in normal people, which may be attributed to DYRK1A. It is reported that DYRK1A is overexpressed in gliomas [81], head and neck squamous cell carcinoma, and non-small cell lung cancer [82]. DYRK1A induces leukemia by phosphorylating NFATc transcription factors and promoting their cytoplasmic retention [23, 83]. The specific mechanism of DYRK1A promoting the occurrence and development of tumors may be attributed to the phosphorylation of cyclin D and enhancement of the P27Kip and the following inhibition of certain transcription factors and the block of cell cycle [84]. By phosphorylating SORY2 and then interfering the ubiquitination of epidermal growth factor receptor (EGFR) mediated by Cbl, DYRK1A promotes the stability of it [85]. And the overexpression of EGFR is generally detected in lung cancer [86-88], glioblastoma [89], breast cancer [90], pancreatic ductal adenocarcinoma [6], esophageal squamous cell carcinoma [91] and many other types of cancer [92]. Therefore, DYRK1A may also promote the occurrence and development of tumors for the involving in the mutation pathway of EGFR (Fig. 1).

|
Download:
|
| Fig. 1. The mechanism of DYRK1A in different diseases (AD, DS, diabetes, cancer). The inhibition of DYRK1A in AD can restore the balance of 3R/4R Tau protein by blocking exons in Tau mRNA, thus promoting Tau aggregation. The selective DYRK1A inhibitor harmine can block the phosphorylation of Ser408 on GLI1, and the activity of mTORC1 is also negatively regulated by DYRK1A. DYRK1A inhibitors can effectively control the development of diabetes by promoting β cell differentiation. DYRK1A may also promote the occurrence and development of tumors for the involving in the mutation pathway of EGFR. | |
{kind=link}
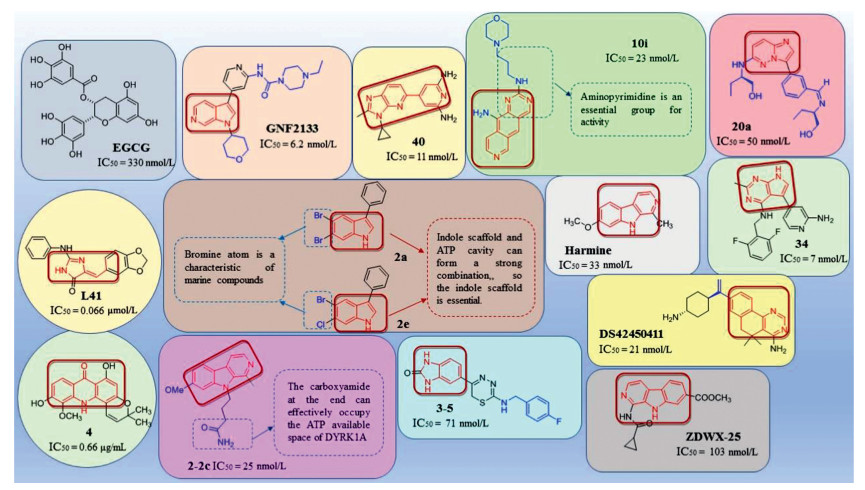
Epigallocatechin-3-gallate (EGCG), a kind of polyphenol found in green tea, can reduce the risk of some neurodegenerative diseases, cardiovascular diseases and even cancer [93-96]. EGCG was originally used as a drug candidate for the treatment of DS for inhibiting DYRK1A in vitro [97, 98]. However, Goodlett et al. [99] used EGCG with different dose for different kinds of mice and found that pure EGCG can reduce the activity of DYRK1A. Unfortunately, EGCG cannot improve the behavior and cognitive deficits of DS mice, and it may carry some dose-related risk for interfering with growth and skeletal integrity (Fig. 2).

|
Download:
|
| Fig. 2. DYRK1A inhibitors. | |
{kind=link}
Recently, it was discovered that DYRK1A is related to regulators in the regenerative pathway, which functions in the secretion of insulin from human pancreatic β cells. And harmine, a potent inhibitor of DYRK1A, is modified to many derivatives for the discovery of anti-diabetes drugs. Binding mode shows that harmine derivatives form two hydrogen bond interactions with Lys188 in the adenosine triphosphate (ATP) pocket of DYRK1A and Leu241 in the side chain of the scaffold [100]. The preservation of 1-methyl and 7-methoxy groups and the replacement of 9-N indole with 9-N glycine can effectively improve the ability of β-cell proliferation, while methyl ester or carboxyamide at the end can effectively occupy the ATP available space of DYRK1A. The chain length between the above two parts is kept at three carbon chain lengths, which have the strongest inhibitory activity on DYRK1A. The length of the three carbon chains can maintain the semi-flexible conformation of the compound, which is beneficial to the binding to the protein. Compound 2-2c (IC50 = 25 nmol/L), a 9-N glycine harmine derivative, has stronger inhibitory activity on DYRK1A than harmine, and can effectively induce NFAT translocation. And most importantly, 2-2c can also induce human β cell proliferation and improve β cell differentiation. The above excellent performance can be attributed to the additional hydrogen bond formed by carboxyamide and Glu291. This additional hydrogen bond may also bring better kinase selectivity to compound 2-2c. Compound 2-2c is of excellent off-target spectrum in vivo and in vitro indicates its good selectivity for DYRK1A. Compound 2-2c can not only increase the β-cell marker (Ki67) but also improve the quality of β cell in vivo. DYRK1A inhibitors have more obstacles as diabetes treatment drugs, such as kinase selectivity as well as off-target effects on the CNS and other tissues (Fig. 2).
4.1.3. Acridone alkaloidsIn recent years, tropical plant extracts have become more and more researched, Beniddir et al. [101] found that the EtOAc bark extract of G. chlorosperma exhibited the most effective inhibitory activity against DYRK1A (IC50 = 0.66 µg/mL). Molecular docking showed that strong, stabilizing interactions of the ligand with both conserved Lys188 and backbone atoms in the hinge region (Glu239 and/or Leu241) are required for good biological activity (e.g., compound 4 and harmine). This is the first report of a natural acridone with potent DYRK1A inhibitory effect, which provides an active scaffold for DYRK1A inhibitors (Fig. 2). And the docking results are consistent with the activity data and provides an explanation for the good inhibitory effect.
4.1.4. LeucettinesDYRK1A plays an important role in DS, specifically, it is located in the DS chromosome 21 critical region which encodes a serine/threonine kinase of numerous substrates. DYRK1A phosphorylates cytoskeletal substrates such as β-tubulin in the cytoplasm, and can also regulate the nuclear/cytoplasmic localization of the NFAT transcription factors. Therefore, when DYRK1A is overexpressed in DS, it can lead to cognitive impairments [23, 102]. To verify the above conclusions, Nguyen et al. [103] treats different mice [Tg(DYRK1A), Ts65Dn and Dp1Yey)] by DYRK1A inhibitor leucine (Leucettine, L41 as an example) and finds that DYRK1A is present in synaptosomes and may affect the transport of synaptosomes, which shows the specific relationship of DYRK1A and recognition memory. L41 can also dephosphorylate SYN1, a protein related to seizures, which broadens the application of DYRK1A inhibitors. Although the specific mechanism of L41 improving the cognitive function of DS patients requires further research, it is undeniable that L41 is a potential drug candidate for the treatment of DS (Fig. 2).
4.1.5. MeridianinsStudies have shown that GSK-3β, CK1δ, CLK1 and DYRK1A act together in the occurrence and development of AD. It has been shown that meridianins, indole alkaloids of the marine tunicate Aplidium from the Southern Ocean, could act as inhibitors of these four kinases. In addition, kororamides A–B, two brominated alkaloids of the bryozoan Amathia tortuosa from Australia, showed a phenotypic signature on Parkinson's disease. Modification of meridianins derivatives mainly focus on the indole scaffold and the substitution of halogen [104]. The ATP cavity of protein kinase can be divided into five regions: glycine-rich region (GRR), hydrophobic pocket (HP), adenine region (AR), sugar pocket (SP) and phosphate binding pocket (PBP) [105-107]. Therefore, the basis for the design of inhibitors is to effectively occupy these cavities. Indole scaffold and ATP cavity can form a strong combination, so the indole scaffold is essential. Compared with terrestrial molecules, marine compounds have at least one bromine atom, which could improve oral absorption, lipophilicity, blood brain barrier (BBB) permeability, metabolic and chemical stability, or even potency. Compounds 2a and 2e have strong binding interactions to protein kinase with generally selective. In addition, the computer predicted that although they had good absorption property and distribution property. But toxic tendencies indicate further modifications are required. All of the above conclusions are based on computer prediction, and the specific parameters of in vitro and in vivo activities need further study (Fig. 2).
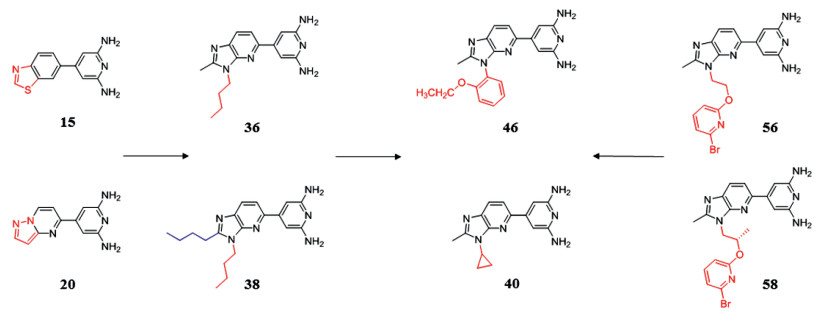
4.2. Synthetic DYRK1A inhibitors 4.2.1. 2, 4′-Bipyridine derivativesThe natural product harmine has been identified as the most effective DYRK1A inhibitor scaffold due to its good selectivity and potent inhibitory effect on DYRK1A [100, 108]. On this basis, Weber et al. [109] developed a variety of new DYRK1A inhibitor scaffolds, which greatly enriched the types of DYRK1A inhibitors. First of all, the selectivity mainly attributes to the interaction between inhibitor and the backbone carbonyl of Leu241. The scaffold was introduced into the thiazole to obtain compound 15 with nanomolar potency (IC50 = 1.0 nmol/L), unfortunately, the sulfur of 15 blocked further growth at this position. But replacing S atom by N atom to form a pyrazole ring (compound 20 IC50 = 2.0 nmol/L) breaks this limitation. The five-membered ring is further expanded into a six-membered ring, which is expected to contact the hinge region sterically accommodated to bring better inhibitory effect. The quinoline derivative and benzodioxane derivatives bring IC50s of 10 and 11 nmol/L, respectively. The above results confirm that the previous design strategy, providing a heteroatom to accept a hydrogen bond from the hinge amide nitrogen of Leu241 to improve selectivity, is correct. More than one amino group on the pyridine is required to form an interaction with the Glu203 to increase the binding affinity to DYRK1A. The addition of the non-polar groups on the five-membered ring seems to maintain a high affinity to DYRK1A, high selectivity versus CDK9, and potent inhibition of DYRK1A autophosphorylation in the cellular setting (36 IC50 = 2 nmol/L, 38 IC50 = 8 nmol/L), which consolidates the choice of the imidazopyridine core for future optimization. The next optimization of inhibitors focused on the improvement of cellular activity. 46 was obtained by introducing 2-PhO-ethyl into imidazole, which provides the highest cellular inhibitory activity (IC50 = 35 nmol/L), and strong inhibitory effect against DYRK1A (IC50 = 1 nmol/L). It is deduced that the 2-PhO-ethyl side chain of 46 sits alongside the imidazo-pyridine core removes this apolar substituent from water, and rigidifies the linker to confine the phenyl to its DYRK1A-bound orientation. Based on this, replacing benzene with pyridine and introducing halogen atoms into pyridine was well tolerated. The compounds with bromine (56) or chlorine (57) was well tolerated by DYRK1A giving 2.0 nmol/L-ar or 3.0 nmol/L-ar inhibitors, respectively. In addition, the cellular most potent inhibitory effect of 56 and 57 were observed at 32 and 26 nmol/L, respectively. In the kinase selectivity experiment, DYRK1A and DYRK2 were inhibited to 10% activity by compound 40 at a lower dose, and 40 induced a 52% decrease of hERG activity at 10 µmol/L. In vitro, 40 have favorable absorption, distribution, metabolism, and excretion (ADME) properties including the complete predicted oral absorption, moderate microsomal stability and a high unbound (free) fraction in plasma. In vivo, the cytostatic effects of 40 on A270 cell line still exist after stopping treating, which suggests that DYRK1A inhibitors, at least in this ovarian model, shows good anti-tumor activity. But it might not be efficient to induce tumor regressions as single agents. Further work is needed to study whether these DYRK1A inhibitors is effective in tumor by combined with other agents (Fig. 3).

|
Download:
|
| Fig. 3. 2, 4'-Bipyridine derivatives. | |
{kind=link}
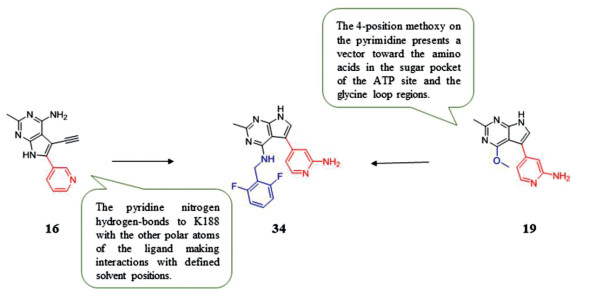
Research shows that DYRK1A and 1B play an important role in cell cycle control and modulation of DYRK1A/B in sarcoma [110, 111], and lung [112], pancreatic [11] and ovarian cancers [113] can result in dysregulation of cell cycle control, and the suppression of DYRK1A restrains myeloid cell leukemia 1 (MCL-1) expression and sensitizes nonsmall cell lung cancer cells to B-cell lymphoma 2 (Bcl-2) inhibitors [114]. Walmsley et al. [115] identified potent and selective ATP competitive inhibitors of DYRK1A and DYRK1B based on the lead discovery method of fragments and structures, which are used to probe inhibitory effect. And they founded that pyrrolopyrimidine compounds can be used for further optimization. The first optimization was concentrated on the carbonyl of L241. Compound 16 (IC50 = 65 nmol/L) was obtained by introduction a methyl group into the 2-position and substitution the 6-position by pyridine, which brings the highest ligand efficiency. The crystal structure shows that the 2-methyl occupies the pocket adjacent to the carbonyl of L241, and the pyridine nitrogen hydrogen-bonds to K188 with the other polar atoms of the ligand making interactions with defined solvent positions. Compound 19 was formed by introducing the methoxy group into pyrrolopyrimidine. Because the methoxy on 4-position of the pyrimidine presents a vector toward the amino acids in the sugar pocket of the ATP site and the glycine loop regions, compound 19 has a high binding affinity for DYRK1A (IC50 = 20 nmol/L). Next, substituents at the 4-position of the pyrimidine aromatic ring was researched, which brings compound 34, the most promising lead compound. Although the metabolic stability is moderate, it shows good cellular activity with the IC50 for inhibition of the autophosphorylation of S520 on DYRK1A in U2OS cells only threefold less than the IC50 in the functional assay and with an concentration for 50% of maximal effect (EC50) of less than 1 nmol/L in a nano-BRET assay. In addition, compound 34 has significant selectivity to DYRK1A (0.4%) and DYRK1B (5%), especially to DYRK2 (88%). It is orally bioavailable and tolerated at doses over 100 mg/kg and achieves sufficient plasma and tumor exposure, showing efficacy in an in vivo model of glioblastoma with a pharmacodynamic (PD) marker (phosphorylation of S520), which confirms cellular target engagement. It is speculated that the methyl group on the 34 pyrropyrimidine occupying a small hydrophobic pocket adjacent to Leu244 to brings good performances. Therefore, the methyl group is a crucial for the selectivity to DYRK1A against other kinases. Compound 34 will be a useful tool to further explore the functional consequence of DYRK1A inhibition which may lead to new therapeutic strategies in oncology and DS (Fig. 4).

|
Download:
|
| Fig. 4. Pyrrolo[2, 3-d]pyrimidine derivatives. | |
{kind=link}
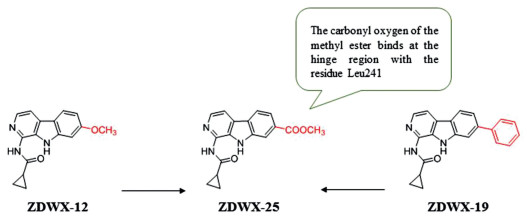
DYRK1A and GSK-3β play a critical role in the occurrence and development of AD. GSK-3β, the main Tau kinase involved in the pathology of AD, is found to be hyperactivated in the brain of AD patients, and DYRK1A is the initiating kinase of GSK-3β signal transduction [116, 117]. Thus, inhibition of GSK-3β and DYRK1A simultaneously will effectively prevent the hyperphosphorylation of Tau, restore the normal function of Tau protein and inhibit the generation of NFTs, which is an encouraging new strategy for anti-AD treatment [118]. Therefore, Liu et al. [119] designed and synthesized harmine derivatives as novel dual GSK 3β/DYRK1A inhibitors based on the β-carboline scaffold. The cyclopropanecarboxamide is linked to different positions of pyridine to form two new types of scaffolds, and the main transformation strategy is concentrated on the 7-position substitution. Among them, the compound ZDWX-12, introducing a methoxy group at the 7 position, has a strong inhibitory activity against DYRK1A (inhibition rate of 85% at 1 µmol/L) and GSK-3β (IC50 = 144 nmol/L). ZDWX-19, substituting the 7-position of phenyl structure by halogen, had moderated GSK-3β inhibitory activity. In addition, compound ZDWX-25 bearing methyl ester at 7-position showed the most promising GSK-3β inhibition activities (IC50 = 71 nmol/L), and also showed the best inhibitory activity against DYRK1A (IC50 = 103 nmol/L) which was at least 450-fold more active than the reference compound harmine. Molecular docking showed that the β-carboline scaffold of the compound ZDWX-25 is well accommodated in the ATP binding pocket of GSK-3β and binds to residues Lys85 and Val135, two crucial amino acids. And, the carbonyl oxygen of the methyl ester binds to the residue Leu241 at the hinge region of DYRK1A, and the nitrogen atom of the pyridine ring forms a hydrogen bond with the side chain of Lys188, which are consistent with the binding mode of the harmine–DYRK1A complex. The above results indicate ZDWX-25 is an ATP-competitive GSK-3β inhibitor. What's important is that ZDWX-25 effectively improved the cognitive dysfunction in APP/PS1/Tau transgenic mice (Fig. 5).

|
Download:
|
| Fig. 5. Pyrido[3, 4-b]indole derivatives. | |
{kind=link}
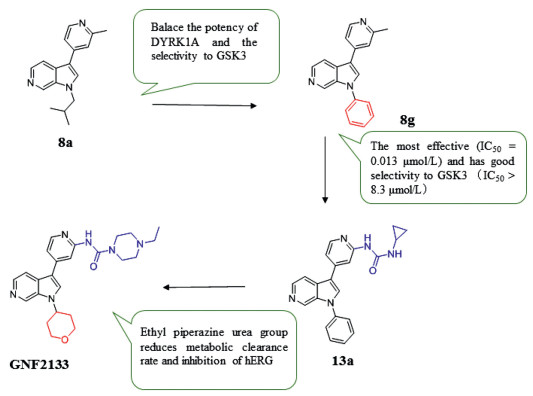
Insufficient β-cells (type 1 diabetes) or restricted β-cell function (type 2 diabetes) are the main pathogenesis of diabetes. Therefore, promoting β cell proliferation and preventing its apoptosis is an effective measure to treat diabetes. Studies have shown that harmine can inhibit the phosphorylation of NFAT in activated T cells to promote the proliferation of β cells by inhibiting DYRK1A in vivo and in vitro. And inhibiting DYRK1A and GSK-3β simultaneously have a synergistic effect on promoting β cell proliferation. Liu et al. [120] explored the groups linked at the nitrogen 1-position of the 6-azaindole core in 8a and substituted by a phenyl group to obtain 8g with good inhibitory effect (IC50 = 0.013 µmol/L) and selectivity to GSK3 (IC50 > 8.3 µmol/L), but the metabolic stability of compound 8g is poor in vivo. It is assumed that substitution at the ortho position of the pyridine nitrogen can reduce the metabolic potential of ADP, which can also form additional hydrogen bond contacts with the scaffold oxygen atom of Leu241. The introduction of urea (compound 13a) displayed higher liver microsomal stability across species, but its inhibition on hERG channel (IC50 = 1.2 µmol/L) brings cardiotoxicity. The ethyl piperazine urea of (GNF2133, compound 13g) contributes to its strong inhibitory effect on DYRK1A (IC50 = 0.0062 µmol/L) and a good selectivity for GSK3, as well as a low metabolic clearance rate and low hERG inhibition. In addition, GNF2133 also shows good proliferation effect on primary β cells and accepted safety in vivo, which confirmed the previous hypothesis that the additional hydrogen bonding contact with the scaffold oxygen atom of Leu241 could increase the potency. 6-Azaindole DYRK1A inhibitor can increase the cell proliferation marker Ki67 and then regulate the cell cycle to influence the proliferation of β cells (Fig. 6).

|
Download:
|
| Fig. 6. Azaindole derivatives. | |
{kind=link}
Studies have shown that all protein kinases share the same co-factor (ATP), and most of their ATP binding pocket are highly conservative in structure, which requires researchers pay more attention on the selectivity of kinase inhibitors [121]. Tazarki et al. [122] synthesized and optimized a series of pyrido[3, 4-g]quinazolines derivatives as inhibitors of Cdc2-like kinase 1 (CLK1) and DYRK1A for their synergistic effect on the occurrence and development of cancer, neurodegenerative diseases and viral infections. The binding modes of pyridoquinazoline derivatives are as follows: the amino group forms a hydrogen bond with Leu244 in the hinge region. And compounds lacking an amino group at position 2 do not show significant inhibition of the tested kinases, which suggests that aminopyrimidine is essential. The compound 10i with an amino group at the 10 position is due to the introduction of the secondary amino group interacts with either the backbone oxygen of Leu244 or points in the direction of the Lys191–Glu206 salt bridge, but without forming additional electrostatic interactions with the latter. Therefore, it has better inhibitory activity on DYRK1A. This article provides a promising scaffold for the development of dual CLK1/DYRK1A inhibitors (Fig. 2).
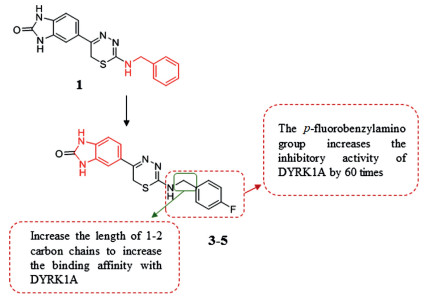
4.2.6. Thiadiazine derivativesIt has been disclosed that DYRK1A plays a key role in cancer. Recent studies have reported that inhibiting DYRK1A may promote EGFR degradation and then inhibit the growth of tumors. Furthermore, DYRK1A inhibition induces activation of caspase-9 which leads to massive apoptosis in specific cancer cell types. Kumar et al. [123] used an integrated computational modeling and medicinal chemistry approach to screen novel DYRK1A inhibitor scaffolds. At first, DFG-in conformation was used to confirm these inhibitors as canonical type-I, ATP competitive inhibitors [12, 124-126]. And Kumar et al. constructed models of DYRK1A in the DFG-out conformation for virtual screening to identify hits with novel binding modes. Notably, gatekeeper residue Phe238 is a key feature of DYRK1A, which is located at the narrow channel bridging the ATP-binding pocket and the DFG-pocket. Screened from the ZINC12 molecular library, 1, 3, 4-thiadiazine compound 1 with good inhibitory effect on DYRK1A (IC50 = 9.41 µmol/L) was discovered. The Kd (71 nmol/L) of the p-fluorobenzylamino introduced at position 2 (compounds 3–5) are of 60 times higher potency than that of compound 1, which indicates that this group is good for the combination of inhibitor and DYRK1A. Increasing the carbon chain length of between the 2-amino group and a phenyl group by 1–2 carbons could increase the binding affinity with DYRK1A. With the above characteristics, compounds 3–5 induced β cell proliferation at 5 µmol/L in vitro, and showed good kinase selectivity for DYRK1A (compared with DYRK1B and DYRK2 with similar structure to DYRK1A). The data all above indicate that compounds 3–5 thiadiazine group can be used as the scaffold for further research and development of DYRK1A inhibitors (Fig. 7).

|
Download:
|
| Fig. 7. Thiadiazine derivatives. | |
{kind=link}
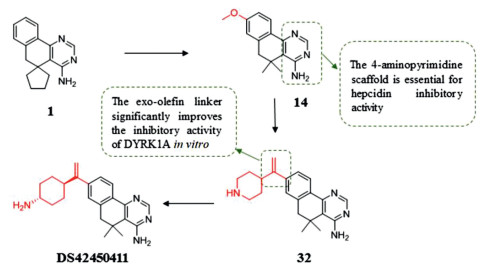
Chronic anemia (ACD) is a common form of anemia only second to iron deficiency anemia [127], which often occurred in patients with acute or chronic immune activation. Hypoxydine underexpression is a common phenotype of hereditary hemochromatosis. Regulation of hepcidin would be a promising treatment strategy for ACD for hepcidin has emerged as the central regulatory molecule in systemic iron homeostasis, and oral hepsidrin inhibitors improve patient compliance. Fukuda et al. [128] hypothesized that the mechanism of inhibition of hepcidin production is due to inhibition of DYRK1A or kinases belonging to CMGC family. Compound 1 (inhibitory activity for hepcidin IC50 = 4.2 µmol/L) with 4-aminopyrimidine was obtained by screening the library of compounds [128]. And the optimization of compound 1 indicates that methoxy group at 8-position can improve the inhibitory effect (compound 14, IC50 = 0.53 µmol/L). When the scaffold was replaced with pyrazole and isoxazole, the activity was found to be lost, which suggests that the 4-aminopyrimidine scaffold is essential for hepcidin inhibitory effect. And the aminopyridine moiety binds to the ATP pocket of DYRK1A by interacting with the hinge region. Compound 32 is obtained by linking the 8-position of piperidine with an exo-olefin linker, which significantly improves the in vitro activity (IC50 = 0.021 µmol/L). This is because that the 8-position NH group of piperidine forms a hydrogen bond with Glu291. In vitro for the NH group at 8-position of piperidine formed a hydrogen bond with the Glu291. In addition, an exo-olefin linker was considered to improve the spatial complementarity with the P-loop. Compound 34 (DS42450411), with similar structure to compound 32, significantly reduces the concentration of hepcidin in the blood. And DS42450411 shows appropriate lipophilicity, moderate plasma protein binding capacity, and high plasma exposure, all of these features meet the original requirements of looking for oral hepcidin inhibitors (Fig. 8).

|
Download:
|
| Fig. 8. Aminopyrimidine derivatives. | |
{kind=link}
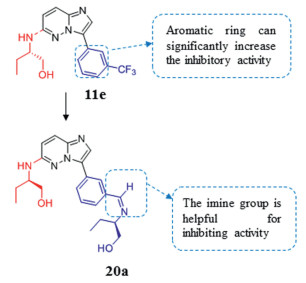
Recent studies have shown that the dysregulation and dysfunction of mammalian CLK and DYRK1A are related to a variety of diseases including single-cell parasitic diseases. Therefore, CLK and DYRK1A have been researched as therapeutic targets. Several patents [129] have described imidazo[1, 2-b]pyridazines as potential antimalarial medicines. The introduction of an aromatic ring at position C-3 greatly improves the activity, and imine is the most active substituent. In addition, compound 11e, substituted by (S)-2-amino-1-butanol at the C-6 position, seems to have the highest activity among a series of compounds introduced with trifluoromethylphenyl (CLK1 IC50 = 0.12 µmol/L). The properties above were combined to obtain the compound 20a, which appears to be the best combination of all tested compounds. Unfortunately, compound 20a cannot inhibit parasites well at a concentration of 10 mmol/L. However, imidazo[1, 2-b] pyridazine scaffold is still a promising candidate for further research as an antimalarial drug (Fig. 9).

|
Download:
|
| Fig. 9. Imidazopyridazine derivatives. | |
{kind=link}
DYRK1A is closely related to many diseases, such as DS and AD. And the correlation between diabetes and DYRK1A is discovered in recent years. Thus, DYRK1A has attracted much attention in drug development. DYRK1A inhibitors must be selective for different diseases, and the pathogenesis of diabetes and neurological diseases are very different. The interaction between DYRK1A inhibitor and protein is mentioned in most articles that DYRK1A inhibitors mainly occupy the ATP binding sites. At present, 3–4 heterocyclic structures are the scaffolds of most DYRK1A inhibitors for better hydrogen bond interaction with proteins, thereby efficiently binding to the DYRK1A domain. Lys188, Leu241, Glu239 and Phe238 are important amino acid residues of DYRK1A, and the compound interact with them has good inhibitory effect, which provides guidance for the design of DYRK1A inhibitors. Zhan et al. [130, 131] proposed that the current design strategies for synthetic compounds mainly include: (1) target-derived (-dependent) de novo drug discovery methodologies, and (2) follow-on derivatization approaches from initially identified active molecules (hit-to-lead and lead-to-candidate efforts). Therefore, the discovery of DYRK1A inhibitors mainly use the second strategy, that is, to design a series of harmine derivatives as DYRK1A inhibitors to explore their therapeutic effects on different diseases, and harmine is a potent inhibitor of DYRK1A. In the process of modification, computer aided design technology is fully utilized. Because the kinases in the DYRK family are similar, the developed inhibitors should be of good selectivity for DYRK1A, which is a big challenge for pharmacologists. In addition to its irreplaceable role in the treatment of DS, AD and diabetes, DYRK1A inhibitors have been found to play a unique role in chronic anemia and osteoarthritis in recent years. Thus, DYRK1A target always attracts attention of researchers.
Declaration of competing interestAll of the authors declare that there is no conflict of interest in connection with this paper.
AcknowledgmentsThis work is funded by the National Natural Science Foundation of China (No. 82073311), National Key Research and Development Program of China (No. 2020YFC2005500), Key Research and Development Program of Science and Technology Department of Sichuan Province (No. 2019YFS0514), Clinical Research and Transformation Fund of Sichuan Provincial People's Hospital (No. 2021LZ03) and the State Administration of Traditional Chinese Medicine (No. JDZX2015210).
| [1] |
S. Gross, R. Rahal, N. Stransky, C. Lengauer, K. Hoeflich, J. Clin. Investig. 125 (2015) 1780-1789. DOI:10.1172/JCI76094 |
| [2] |
A. Zs, A. Ql, M. Lin, Chin. Chem. Lett. 31 (2020) 1345-1356. DOI:10.1016/j.cclet.2020.03.001 |
| [3] |
L. Chico, L. Van Eldik, D. Watterson, Nat. Rev. Drug Discov. 8 (2009) 892-909. DOI:10.1038/nrd2999 |
| [4] |
S. Sharma, J. Singh, R. Ojha, et al., Eur. J. Med. Chem. 112 (2016) 298-346. DOI:10.1016/j.ejmech.2016.02.018 |
| [5] |
Y. Gwack, S. Sharma, J. Nardone, et al., Nature 441 (2006) 646-650. DOI:10.1038/nature04631 |
| [6] |
J. Luna, J. Boni, M. Cuatrecasas, et al., Gut 68 (2019) 1465-1476. DOI:10.1136/gutjnl-2018-316128 |
| [7] |
M. Soundararajan, A. Roos, P. Savitsky, et al., Structure 21 (2013) 986-996. DOI:10.1016/j.str.2013.03.012 |
| [8] |
S. Aranda, A. Laguna, S. de la Luna, FASEB J. 25 (2011) 449-462. DOI:10.1096/fj.10-165837 |
| [9] |
S.B. Lee, A. Ko, Y.T. Oh, et al., Mol. Cell 79 (2020) 376-389.e378. DOI:10.1016/j.molcel.2020.06.021 |
| [10] |
J. Gao, X. Yang, P. Yin, et al., Int. J. Oncol. 40 (2012) 1203-1209. DOI:10.3892/ijo.2011.1293 |
| [11] |
X. Deng, D. Ewton, S. Li, et al., Cancer Res. 66 (2006) 4149-4158. DOI:10.1158/0008-5472.CAN-05-3089 |
| [12] |
Y. Ogawa, Y. Nonaka, T. Goto, et al., Nat. Commun. 1 (2010) 86. DOI:10.1038/ncomms1090 |
| [13] |
B. van Bon, B. Coe, R. Bernier, et al., Mol. Psychiatry 21 (2016) 126-132. DOI:10.1038/mp.2015.5 |
| [14] |
W. Song, E. Song, S. Choi, et al., Neurosci. Lett. 554 (2013) 135-140. DOI:10.1016/j.neulet.2013.08.066 |
| [15] |
J. Park, J. Sung, J. Park, et al., J. Cell Sci. 125 (2012) 67-80. DOI:10.1242/jcs.086124 |
| [16] |
F. Liu, Z. Liang, J. Wegiel, et al., FASEB J. 22 (2008) 3224-3233. DOI:10.1096/fj.07-104539 |
| [17] |
I. Kii, Y. Sumida, T. Goto, et al., Nat. Commun. 7 (2016) 11391. DOI:10.1038/ncomms11391 |
| [18] |
M. Arbones, A. Thomazeau, A. Nakano-Kobayashi, M. Hagiwara, J. Delabar, Pharmacol. Ther. 194 (2019) 199-221. DOI:10.1016/j.pharmthera.2018.09.010 |
| [19] |
T. Dang, W. Duan, B. Yu, et al., Mol. Psychiatry 23 (2018) 747-758. DOI:10.1038/mp.2016.253 |
| [20] |
A. Hz, A. Yq, B. Zz, et al., Chin. Chem. Lett. 32 (2021) 1857-1868. DOI:10.1016/j.cclet.2021.01.014 |
| [21] |
D. Xue, H. Liu, S. Chen, et al., Chin. Chem. Lett. 28 (2017) 1190-1193. DOI:10.1016/j.cclet.2017.03.040 |
| [22] |
J. Wegiel, C. Gong, Y. Hwang, FEBS J. 278 (2011) 236-245. DOI:10.1111/j.1742-4658.2010.07955.x |
| [23] |
J. Arron, M. Winslow, A. Polleri, et al., Nature 441 (2006) 595-600. DOI:10.1038/nature04678 |
| [24] |
R. Kimura, K. Kamino, M. Yamamoto, et al., Hum. Mol. Genet. 16 (2007) 15-23. DOI:10.1093/hmg/ddl437 |
| [25] |
P. Wang, J. Alvarez-Perez, D. Felsenfeld, et al., Nat. Med. 21 (2015) 383-388. DOI:10.1038/nm.3820 |
| [26] |
W. Shen, B. Taylor, Q. Jin, et al., Nat. Commun. 6 (2015) 8372. DOI:10.1038/ncomms9372 |
| [27] |
W. Becker, U. Soppa, F. Tejedor, CNS Neurol. Disord. Drug Targets 13 (2014) 26-33. DOI:10.2174/18715273113126660186 |
| [28] |
B. Smith, F. Medda, V. Gokhale, T. Dunckley, C. Hulme, ACS Chem. Neurosci. 3 (2012) 857-872. DOI:10.1021/cn300094k |
| [29] |
A. Ionescu, F. Dufrasne, M. Gelbcke, et al., Mini Rev. Med. Chem. 12 (2012) 1315-1329. |
| [30] |
P. Fernández-Martínez, C. Zahonero, P. Sánchez-Gómez, Mol. Cell. Oncol. 2 (2015) e970048. DOI:10.4161/23723548.2014.970048 |
| [31] |
F. Tejedor, B. Hämmerle, FEBS J. 278 (2011) 223-235. DOI:10.1111/j.1742-4658.2010.07954.x |
| [32] |
T. Nguyen, C. Fruit, Y. Hérault, L. Meijer, T. Besson, Expert Opin. Ther. Pat. 27 (2017) 1183-1199. DOI:10.1080/13543776.2017.1360285 |
| [33] |
U. Soppa, J. Schumacher, V.F. Ortiz, et al., Cell Cycle 13 (2014) 2084-2100. DOI:10.4161/cc.29104 |
| [34] |
N. Kurabayashi, M. Nguyen, K. Sanada, EMBO Rep. 16 (2015) 1548-1562. DOI:10.15252/embr.201540374 |
| [35] |
B. Khor, J. Gagnon, G. Goel, et al., Elife 4 (2015) e05920. DOI:10.7554/eLife.05920 |
| [36] |
K. Ori-McKenney, R. McKenney, H. Huang, et al., Neuron 90 (2016) 551-563. DOI:10.1016/j.neuron.2016.03.027 |
| [37] |
C. Grau, K. Arató, J. Fernández-Fernández, et al., Front. Cell. Neurosci. 8 (2014) 331. |
| [38] |
B. Belgardt, E. Lammert, Diabetes 65 (2016) 1496-1498. DOI:10.2337/dbi16-0013 |
| [39] |
D. Allsop, J. Mayes, Essays Biochem. 56 (2014) 99-110. DOI:10.1042/bse0560099 |
| [40] |
M. Murphy, H. LeVine, J. Alzheimers Dis. 19 (2010) 311-323. DOI:10.3233/JAD-2010-1221 |
| [41] |
G. Chen, T. Xu, Y. Yan, et al., Acta Pharmacol. Sin. 38 (2017) 1205-1235. DOI:10.1038/aps.2017.28 |
| [42] |
I. Blasko, M. Stampfer-Kountchev, P. Robatscher, et al., Aging Cell 3 (2004) 169-176. DOI:10.1111/j.1474-9728.2004.00101.x |
| [43] |
I. Blasko, F. Marx, E. Steiner, T. Hartmann, B. Grubeck-Loebenstein, FASEB J. 13 (1999) 63-68. DOI:10.1096/fasebj.13.1.63 |
| [44] |
H. Lee, H. Woo, H. Lee, et al., Free Radic. Biol. Med. 160 (2020) 575-595. DOI:10.1016/j.freeradbiomed.2020.08.030 |
| [45] |
R. Medeiros, M. Kitazawa, M. Chabrier, et al., Am. J. Pathol. 181 (2012) 616-625. DOI:10.1016/j.ajpath.2012.04.020 |
| [46] |
P. Rudrabhatla, H. Jaffe, H. Pant, FASEB J. 25 (2011) 3896-3905. DOI:10.1096/fj.11-181297 |
| [47] |
Y. Woods, P. Cohen, W. Becker, et al., Biochem. J. 355 (2001) 609-615. DOI:10.1042/bj3550609 |
| [48] |
S. Ryoo, H. Cho, H. Lee, H. Kyeong, J. Chinzorig, J. Neurochem. 104 (2008) 1333-1344. DOI:10.1111/j.1471-4159.2007.05075.x |
| [49] |
Y. Ryu, S. Park, M. Jung, et al., J. Neurochem. 115 (2010) 574-584. DOI:10.1111/j.1471-4159.2010.06769.x |
| [50] |
B. Hämmerle, C. Elizalde, F. Tejedor, Eur. J. Neurosci. 27 (2008) 1061-1074. DOI:10.1111/j.1460-9568.2008.06092.x |
| [51] |
S. Darwish, M. Abdel-Halim, A. ElHady, et al., Eur. J. Med. Chem. 158 (2018) 270-285. DOI:10.1016/j.ejmech.2018.08.097 |
| [52] |
K. Iqbal, F. Liu, C. Gong, Nat. Rev. Neurol. 12 (2016) 15-27. DOI:10.1038/nrneurol.2015.225 |
| [53] |
C. Branca, D. Shaw, R. Belfiore, et al., Aging Cell 16 (2017) 1146-1154. DOI:10.1111/acel.12648 |
| [54] |
D. Frost, B. Meechoovet, T. Wang, et al., PLoS One 6 (2011) e19264. DOI:10.1371/journal.pone.0019264 |
| [55] |
P. Chang, D. Bush, S. Schorge, et al., Cell Rep. 30 (2020) 1152-1163.e1154. DOI:10.1016/j.celrep.2019.12.065 |
| [56] |
P. Schneider, J. Bayo-Fina, R. Singh, et al., Nat. Commun. 6 (2015) 8023. DOI:10.1038/ncomms9023 |
| [57] |
A. Mégarbané, A. Ravel, C. Mircher, et al., Genet. Med. 11 (2009) 611-616. DOI:10.1097/GIM.0b013e3181b2e34c |
| [58] |
K. Gardiner, Trends Pharmacol. Sci. 31 (2010) 66-73. DOI:10.1016/j.tips.2009.10.010 |
| [59] |
J. Jiang, Y. Jing, G. Cost, et al., Nature 500 (2013) 296-300. DOI:10.1038/nature12394 |
| [60] |
K. Baek, A. Zaslavsky, R. Lynch, et al., Nature 459 (2009) 1126-1130. DOI:10.1038/nature08062 |
| [61] |
J. Grieco, M. Pulsifer, K. Seligsohn, B. Skotko, A. Schwartz, Am. J. Med. Genet. C Semin. Med. Genet. 169 (2015) 135-149. DOI:10.1002/ajmg.c.31439 |
| [62] |
I. Lott, M. Dierssen, Lancet Neurol. 9 (2010) 623-633. DOI:10.1016/S1474-4422(10)70112-5 |
| [63] |
C. Clark, F. Fernandez, S. Sakhon, G. Spanò, J. Edgin, Hippocampus 27 (2017) 683-691. DOI:10.1002/hipo.22724 |
| [64] |
P. Lavenex, M. Bostelmann, C. Brandner, et al., Front. Psychol. 6 (2015) 62. |
| [65] |
S. Lanfranchi, O. Jerman, E. Dal Pont, A. Alberti, R. Vianello, J. Intellect. Disabil. Res. 54 (2010) 308-319. DOI:10.1111/j.1365-2788.2010.01262.x |
| [66] |
B. Pennington, J. Moon, J. Edgin, J. Stedron, L. Nadel, Child Dev. 74 (2003) 75-93. DOI:10.1111/1467-8624.00522 |
| [67] |
J. Rowe, A. Lavender, V. Turk, Br. J. Clin. Psychol. 45 (2006) 5-17. DOI:10.1348/014466505X29594 |
| [68] |
J. Anderson, J. Nielsen, M. Ferguson, et al., Neuroimage Clin. 2 (2013) 703-715. DOI:10.1016/j.nicl.2013.05.006 |
| [69] |
M. Ruiz-Mejias, M. Martinez de Lagran, M. Mattia, et al., J. Neurosci. 36 (2016) 3648-3659. DOI:10.1523/JNEUROSCI.2517-15.2016 |
| [70] |
A. Duchon, Y. Herault, Front. Behav. Neurosci. 10 (2016) 104. DOI:10.3389/fnbeh.2016.00104 |
| [71] |
R. Roper, L. Baxter, N. Saran, et al., Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 1452-1456. DOI:10.1073/pnas.0510750103 |
| [72] |
J. Mao, P. Maye, P. Kogerman, et al., J. Biol. Chem. 277 (2002) 35156-35161. DOI:10.1074/jbc.M206743200 |
| [73] |
T. Shimokawa, U. Tostar, M. Lauth, et al., J. Biol. Chem. 283 (2008) 14345-14354. DOI:10.1074/jbc.M800299200 |
| [74] |
L. Larsen, L. Møller, Cells 9 (2020) 2316. DOI:10.3390/cells9102316 |
| [75] |
B. Ehe, D. Lamson, M. Tarpley, et al., Biochem. Biophys. Res. Commun. 491 (2017) 767-772. DOI:10.1016/j.bbrc.2017.07.107 |
| [76] |
B. Ehe, D. Lamson, M. Tarpley, et al., Data Brief 15 (2017) 577-583. DOI:10.1016/j.dib.2017.09.057 |
| [77] |
D. Robbins, D. Fei, N. Riobo, Sci. Signal. 5 (2012) re6. DOI:10.1126/scisignal.2002906 |
| [78] |
B. Stecca, A. Ruiz i Altaba, J. Neurobiol. 64 (2005) 476-490. DOI:10.1002/neu.20160 |
| [79] |
X. Chen, J. Fu, F. Zhou, Q. Yang, J. Wang, et al., J. Med. Chem. 63 (2020) 12595-12613. DOI:10.1021/acs.jmedchem.0c00736 |
| [80] |
P. Wang, N. Fiaschi-Taesch, R. Vasavada, et al., Nat. Rev. Endocrinol. 11 (2015) 201-212. DOI:10.1038/nrendo.2015.9 |
| [81] |
N. Pozo, C. Zahonero, P. Fernández, et al., J. Clin. Investig. 123 (2013) 2475-2487. DOI:10.1172/JCI63623 |
| [82] |
A. Radhakrishnan, V. Nanjappa, R. Raja, et al., Sci. Rep. 6 (2016) 36132. DOI:10.1038/srep36132 |
| [83] |
S. Malinge, M. Bliss-Moreau, G. Kirsammer, et al., J. Clin. Investig. 122 (2012) 948. DOI:10.1172/JCI60455 |
| [84] |
A. Laham, M. Saber-Ayad, R. El-Awady, Cell. Mol. Life Sci. 78 (2021) 603-619. DOI:10.1007/s00018-020-03626-4 |
| [85] |
X. Xu, Q. Liu, C. Zhang, et al., Cell Death Dis. 10 (2019) 282. DOI:10.1080/15476286.2019.1572435 |
| [86] |
L. Ding, G. Getz, D. Wheeler, et al., Nature 455 (2008) 1069-1075. DOI:10.1038/nature07423 |
| [87] |
R. Rosell, T. Moran, C. Queralt, et al., N. Engl. J. Med. 361 (2009) 958-967. DOI:10.1056/NEJMoa0904554 |
| [88] |
M. Imielinski, A. Berger, P. Hammerman, et al., Cell 150 (2012) 1107-1120. DOI:10.1016/j.cell.2012.08.029 |
| [89] |
C. Brennan, R. Verhaak, A. McKenna, et al., Cell 155 (2013) 462-477. DOI:10.1016/j.cell.2013.09.034 |
| [90] |
J. Kim, A. Siverly, D. Chen, et al., Cancer Res. 76 (2016) 6424-6435. DOI:10.1158/0008-5472.CAN-16-1571 |
| [91] |
Y. Song, L. Li, Y. Ou, et al., Nature 509 (2014) 91-95. DOI:10.1038/nature13176 |
| [92] |
N. Normanno, A. De Luca, C. Bianco, et al., Gene 366 (2006) 2-16. DOI:10.1016/j.gene.2005.10.018 |
| [93] |
S. Mandel, T. Amit, L. Reznichenko, O. Weinreb, M. Youdim, Mol. Nutr. Food Res. 50 (2006) 229-234. DOI:10.1002/mnfr.200500156 |
| [94] |
A. Chowdhury, J. Sarkar, T. Chakraborti, P. Pramanik, S. Chakraborti, Biomed. Pharmacother. 78 (2016) 50-59. DOI:10.1016/j.biopha.2015.12.013 |
| [95] |
M. Cascella, S. Bimonte, M. Muzio, V. Schiavone, A. Cuomo, Infect. Agents Cancer 12 (2017) 36. DOI:10.1186/s13027-017-0145-6 |
| [96] |
B. Rashidi, M. Malekzadeh, M. Goodarzi, A. Masoudifar, H. Mirzaei, Biomed. Pharmacother. 89 (2017) 949-956. DOI:10.1016/j.biopha.2017.01.161 |
| [97] |
J. Bain, H. McLauchlan, M. Elliott, P. Cohen, Biochem. J. 371 (2003) 199-204. DOI:10.1042/bj20021535 |
| [98] |
T. Adayev, M. Chen-Hwang, N. Murakami, et al., Biochemistry 45 (2006) 12011-12019. DOI:10.1021/bi060632j |
| [99] |
C. Goodlett, M. Stringer, J. LaCombe, et al., Sci. Rep. 10 (2020) 10426. DOI:10.1038/s41598-020-67133-z |
| [100] |
K. Kumar, P. Wang, J. Wilson, et al., J. Med. Chem. 63 (2020) 2986-3003. DOI:10.1021/acs.jmedchem.9b01379 |
| [101] |
M. Beniddir, E.Le Borgne, B. Iorga, et al., J. Nat. Prod. 77 (2014) 1117-1122. DOI:10.1021/np400856h |
| [102] |
M. Alvarez, X. Altafaj, S. Aranda, S. de la Luna, Mol. Biol. Cell 18 (2007) 1167-1178. DOI:10.1091/mbc.e06-08-0668 |
| [103] |
T. Nguyen, A. Duchon, A. Manousopoulou, et al., Dis. Models Mech. 11 (2018) 035634 dmm035634. |
| [104] |
L. Llorach-Pares, A. Nonell-Canals, C. Avila, M. Sanchez-Martinez, Mar. Drugs 16 (2018) 386. DOI:10.3390/md16100386 |
| [105] |
P. Traxler, P. Furet, Pharmacol. Ther. 82 (1999) 195-206. DOI:10.1016/S0163-7258(98)00044-8 |
| [106] |
M. McGregor, J. Chem. Inf. Model. 47 (2007) 2374-2382. DOI:10.1021/ci700244t |
| [107] |
D. Huang, T. Zhou, K. Lafleur, C. Nevado, A. Caflisch, Bioinformatics 26 (2010) 198-204. DOI:10.1093/bioinformatics/btp650 |
| [108] |
J. Bain, L. Plater, M. Elliott, et al., Biochem. J. 408 (2007) 297-315. DOI:10.1042/BJ20070797 |
| [109] |
C. Weber, M. Sipos, A. Paczal, et al., J. Med. Chem. 64 (2021) 6745-6764. DOI:10.1021/acs.jmedchem.1c00023 |
| [110] |
S. Mercer, D. Ewton, S. Shah, A. Naqvi, E. Friedman, Cancer Res. 66 (2006) 5143-5150. DOI:10.1158/0008-5472.CAN-05-1539 |
| [111] |
C. Yang, D. Ji, E. Weinstein, et al., Carcinogenesis 31 (2010) 552-558. DOI:10.1093/carcin/bgp330 |
| [112] |
J. Gao, Z. Zheng, B. Rawal, et al., Cancer Biol. Ther. 8 (2009) 1671-1679. DOI:10.4161/cbt.8.17.9322 |
| [113] |
J. Hu, H. Deng, E.A. Friedman, Int. J. Cancer 132 (2013) 2258-2269. DOI:10.1002/ijc.27917 |
| [114] |
Y. Li, D. Zhou, S. Xu, et al., Cancer Biol. Med. 17 (2020) 387-400. DOI:10.20892/j.issn.2095-3941.2019.0380 |
| [115] |
D.Lee Walmsley, J. Murray, P. Dokurno, et al., J. Med. Chem. 64 (2021) 8971-8991. DOI:10.1021/acs.jmedchem.1c00024 |
| [116] |
K. Patel, M. Gadewar, R. Tripathi, S. Prasad, D. Patel, Asian Pac. J. Trop. Biomed. 2 (2012) 660-664. DOI:10.1016/S2221-1691(12)60116-6 |
| [117] |
H. Song, Y. Liu, Y. Liu, L. Wang, Q. Wang, J. Agric. Food Chem. 62 (2014) 1010-1018. DOI:10.1021/jf404840x |
| [118] |
C. Labrière, O. Lozach, M. Blairvacq, L. Meijer, C. Guillou, Eur. J. Med. Chem. 124 (2016) 920-934. DOI:10.1016/j.ejmech.2016.08.069 |
| [119] |
W. Liu, X. Liu, L. Tian, et al., Eur. J. Med. Chem. 222 (2021) 113554. DOI:10.1016/j.ejmech.2021.113554 |
| [120] |
Y. Liu, Q. Jin, Y. Zou, et al., J. Med. Chem. 63 (2020) 2958-2973. DOI:10.1021/acs.jmedchem.9b01624 |
| [121] |
S. Müller, A. Chaikuad, N. Gray, S. Knapp, Nat. Chem. Biol. 11 (2015) 818-821. DOI:10.1038/nchembio.1938 |
| [122] |
H. Tazarki, W. Zeinyeh, Y. Esvan, et al., Eur. J. Med. Chem. 166 (2019) 304-317. DOI:10.1016/j.ejmech.2019.01.052 |
| [123] |
K. Kumar, P. Man-Un Ung, P. Wang, et al., Eur. J. Med. Chem. 157 (2018) 1005-1016. DOI:10.1016/j.ejmech.2018.08.007 |
| [124] |
K. Anderso, Y. Chen, Z. Chen, et al., Bioorg. Med. Chem. Lett. 23 (2013) 6610-6615. DOI:10.1016/j.bmcl.2013.10.055 |
| [125] |
H. Falke, A. Chaikuad, A. Becker, N. Loac, C. Kunick, J. Med. Chem.. DOI:10.1021/jm501994d |
| [126] |
U. Rothweiler, W. Stensen, B. Brandsdal, et al., J. Med. Chem. 59 (2016) 9814-9824. DOI:10.1021/acs.jmedchem.6b01086 |
| [127] |
G. Weiss, L. Goodnough, N. Engl. J. Med. 352 (2005) 1011-1023. DOI:10.1056/NEJMra041809 |
| [128] |
T. Fukuda, T. Ishiyama, T. Katagiri, et al., Bioorg. Med. Chem. Lett. 28 (2018) 3333-3337. DOI:10.1016/j.bmcl.2018.09.010 |
| [129] |
L. Bendjeddou, N. Loaëc, B. Villiers, et al., Eur. J. Med. Chem. 125 (2017) 696-709. DOI:10.1016/j.ejmech.2016.09.064 |
| [130] |
P. Zhan, Y. Itoh, T. Suzuki, X. Liu, J. Med. Chem. 58 (2015) 7611-7633. DOI:10.1021/acs.jmedchem.5b00229 |
| [131] |
J. Du, J. Guo, D. Kang, et al., Chin. Chem. Lett. 31 (2020) 1695-1708. DOI:10.1016/j.cclet.2020.03.028 |