 2022, Vol. 33
2022, Vol. 33 


The sufficient utilization of methane (CH4) has been a hot topic for the chemical and energy industries; however, it remains a challenge. Because the C-H bond in methane is the most inert (104 kcal/mol) of all hydrocarbons [1], with low electron affinity and high ionization potential, the high temperature required to activate the C-H bond often leads to methane over oxidation to CO2. Methane reserves are usually located in remote areas and are difficult for transportation. In addition, methane extracted during oil and gas production is often flared to avoid emission of this potent greenhouse gas into the atmosphere. The world bank estimates that about 142 billion cubic meters of methane is flared annually around the world, contributing to about 1.0% of global CO2 emission [2]. Moreover, CO2 can linger in the atmosphere for hundreds of years compared to ~10 years of CH4, and thus the current CH4 flaring to CO2 in the oil production industry is only a short-term solution to reduce the greenhouse effect. Even with many efforts having been made for transforming CO2 into useful chemicals, the ideal solution is transforming methane to liquid oxygenates rather than releasing CO2 [3-6]. Traditionally, methanol (CH3OH) production from methane can be done in two steps, in which methane is firstly converted to syngas, and then syngas is used to produce methanol. However, the two-step syngas methanol production route from methane is energy intensive and only economically feasible at large scale. For the methane produced during oil production, the best way to utilize it is to do a one-step oxidation to methanol under mild conditions. It is challenging to produce methanol from direct methane oxidation since product methanol is more susceptible to oxidation than reactant methane. Expensive or toxic oxidants have been used to resolve this issue but limit its commercial application [7-9]. Using a low-cost oxidant, such as O2, is attractive, however, methane suffers from overoxidation to CO2. Zeolite supported Cu or Fe catalysts can do gas phase oxidation of methane but requires oxidant activation, methanol desorption at high temperature (200-500 ℃), and shows extreme low reactivity.
In contrast with gas-phase methane oxidation, methane oxidation in the liquid phase is less energy intensive. A catalytic system containing Pt and Hg can oxidize methane to methyl bisulfate, but the methanol production from subsequent hydrolysis generates SO2 [10]. Similarly, cationic Pt, Pd, and Au catalysts can oxidize methane with the use of strong oxidizing agents, such as selenic acid, producing environmentally unfriendly byproducts [10, 11]. To overcome the environmental issues, H2O2 has been used as green oxidant and exhibits high activity and selectivity to CH3OH for methane partial oxidation near ambient temperatures [12-18]; however, it is not economically feasible since H2O2 is more expensive than CH3OH. Many catalysts have been developed for H2O2 production with H2 and O2 in the liquid phase at low temperatures, such as Pd based catalysts [19-23], which offer an alternative approach to partially oxidize CH4. Therefore, developing a catalytic system that can in situ produce H2O2 from H2 and O2 for methane oxidation has been carried out [24, 25]. However, a low methanol yield is often observed, which might be due to the limited amount of H2O2 accessible to the active sites for methane partial oxidation.
Zeolitic materials exhibit better performance for Cu, Fe, and Pd based catalysts due to their pore structure that helps to stabilize active metal species. However, it is well known that expensive and toxic ligands and organic solvents are needed for zeolite synthesis. Aiming to achieve a green catalytic process, it is vital to make the catalyst preparation process green too. In this review, the first section will cover the green oxidant for low temperature methane oxidation, and the second section will cover green approach about zeolite synthesis.
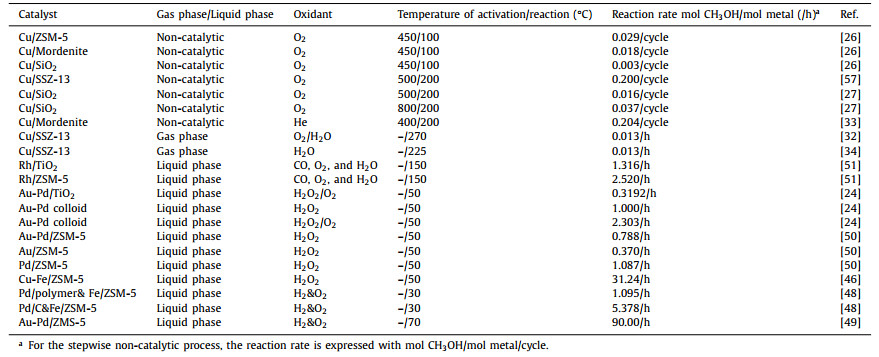
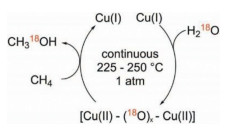
2. CH4 partial oxidation to CH3OH with green oxidants 2.1. Direct use of O2/H2O oxidant for methane partial oxidationO2 is an abundant and cheap oxidant and widely used for numerous reactions. A body of efforts have been made on Cu-zeolites system for methane partial oxidation since the first report found that Cu-zeolites (small pores) were much more active than its counterpart, Cu-SiO2 mixed with Al2O3 [26]; nonetheless, it is not a catalytic, but stoichiometric reaction. Bozbag et al. reported that with solely SiO2 support (not zeolitic), Cu-SiO2 can activate O2 at high temperature and the concurrent Cu dispersion was observed [27]. The higher temperature (within the temperature range investigated, ≤ 800 ℃) of O2 activation over Cu/SiO2, the more active Cu sites formed. The results indicate that the methanol production relates to the Cu oxo active species. However, the process is stepwise and not catalytic, involving the steps of O2 activation, CH4 activation, and CH3OH extraction by H2O (Fig. 1). In addition, Yang et al. have employed CuO as the O provider and the other metals (such as Ir, Ru) to activate CH4 to form CH3OH, providing a new angle to develop highly efficient catalysts for methane oxidation to methanol in a stepwise reaction [28-30]. Moreover, it is not feasible to use high temperature for the continuous catalytic process due to methane overoxidation to CO2. Continuous production from catalytic methane oxidation with O2 and H2O at relatively low temperatures (210-225 ℃) over Cu exchanged zeolites (ZSM-5, mordenite, and SSZ-13) have been reported [31]. This work demonstrates that the continuous catalytic production of methanol from methane at low temperature is possible even the reactivity is extremely low. In contrast, CuOx supported by these zeolites with wetness impregnation method do not show observable activity under the same conditions, which confirms the Cu oxo species are active sites.

|
Download:
|
| Fig. 1. Graphic representation of the stepwise reaction steps for methane partial oxidation and the O2 activation temperature vs methanol yield. Reproduced with permission [27]. Copyright 2018, American Chemical Society. | |
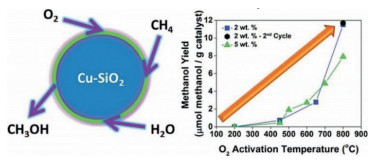
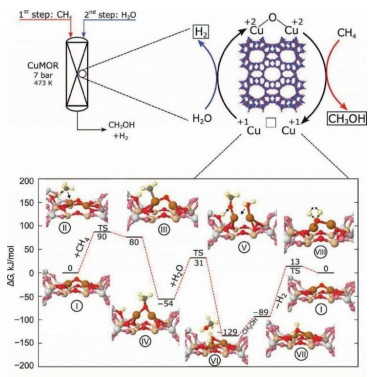
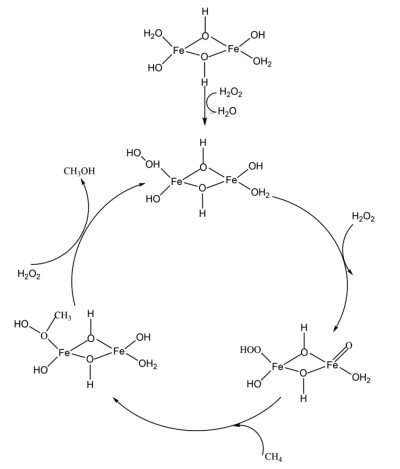
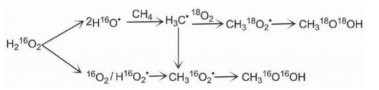
H2O can work as the oxidant for many reactions and it is frequently applied to methane oxidation to methanol system and it has been shown to be able to extract methanol from the catalyst surface. Moreover, Kimberly et al. found that C1 intermediate on [Cu-O2-Cu]2+ desorbs as methanol with the presence of H2O/H+, leading to methanol production or completely oxidized to CO2 by O2 [32]. Interestingly, Sushkevich et al. found that H2O itself can oxidize methane to methanol continuously over Cu exchanged ZSM-5 with limited reactivity. The role of water was hypothesized as replenishing the bridge O in Cu-O2-Cu in the catalytic cycle with concurrent H2 production (Fig. 2) [33]. Koishybay et al. also found that water could oxidize CH4 to methanol in a continuous catalytic fashion and the O in methanol originates from H2O as demonstrated by isotopic experiment and the molecular reaction mechanism was indicated in Fig. 3 [34]. Moreover, by using reducible CeO2/Cu2O on Cu(111) surface, Liu et al. found that H2O preferentially adsorbed on CeO2 and dissociated into a hydroxyl that promotes the oxidation of CH4 to CH3OH and H2O and also blocks O-O dissociation that leads to CO and CO2 from CH4. Subsequently, O2 helps to oxidize the reduced CeOx back to CeO2 [35].

|
Download:
|
| Fig. 2. Scheme of reaction mechanism of methane oxidation solely with H2O oxidant with concurrent production of H2 and CH3OH. Reproduced with permission [33]. Copyright 2017, American Association for the Advancement of Science. | |

|
Download:
|
| Fig. 3. Proposed reaction mechanism for methane oxidation by water over Cu/SSZ-13. Reproduced with permission [34]. Copyright 2020, American Chemical Society. | |
Since activation of O2 for methane partial oxidation requires high temperature that leads to high energy input and poor selectivity towards oxygenate products. To activate methane for oxygenate products under mild conditions, strong oxidants such as SO3 [9, 36, 37], and K2SO3 [9, 38-43] are usually needed. For example, CH4 can be oxidized by SO3 via reaction (1) to a valuable chemical CH3SO3H (methane sulfonic acid).
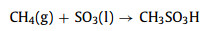
|
(1) |
Besides strong oxidants; H2O2 has also been used for CH4 oxidation under mild conditions. 10% methane conversion and 90% methanol selectivity were achieved over Fe-ZMS-5 catalyst by using H2O2 as an oxidant [13], in which H2O2 plays a critical role in activating the iron species that activates the C-H bond and converts methane to oxygenates (Fig. 4). In other catalytic systems, support free Au-Pd colloids, have shown the ability to catalyze methane to methanol with both H2O2 and O2, and the role of H2O2 was suggested to be activating CH4 to ·CH3 radical followed by O2 insertion to produce methyl hydroperoxide (CH3OOH) that can readily transform to CH3OH (Fig. 5). H2O2 can be used as the oxidant for methane oxidation over various catalytic system Cu/C3N4 [12], Fe/ZSM-5 [13], Cr/TiO2 [14], Pd/ZSM-5 [15], Ni/CNT-NC (N doped carbon) [16], Ir/TiO2 [17], Rh/ZrO2 [18], etc. H2O2 works well to oxidize CH4 to methanol; however, its high cost creates hurdles for the potential application.

|
Download:
|
| Fig. 4. Proposed methane oxidation reaction mechanism with H2O2 oxidant for Fe based catalyst [12]. Reproduced with permission [12]. Copyright 2012, John Wiley & Sons. | |

|
Download:
|
| Fig. 5. Proposed reaction network for methane oxidation with H2O2 and O2 over Au-Pd catalyst. Reproduced with permission [24]. Copyright 2017, American Association for the Advancement of Science. | |
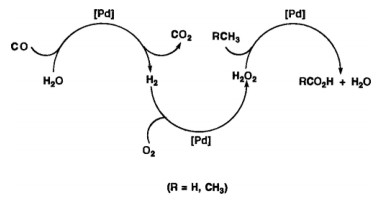
To overcome this, in situ production of H2O2 from O2 and H2 for methane oxidation seems a promising approach since numerous reports of H2O2 catalytic production from H2 and O2 over various catalytic systems [19, 21, 23, 44-46]. Therefore, in situ generated H2O2 using H2 or (CO) in solution has been attempted for methane oxidation to methanol [25, 47-51]. CO was used as the reductive promoter to activate O2 for methane oxidation. With metallic Pd and CuCl2 in solution [52], CO was found to generate H2 via water gas shift reaction (CO + H2O → H2 + CO2) over Pd site, and the produced H2 reacts with O2 to generate H2O2, which subsequently activates CH4 to produce methanol and acetic acid (Scheme 1). Moreover, Shan et al. used CO to activate a CH4 molecule for the methane oxidation reaction over Rh/ZSM-5, and got the product distribution of methanol, acetic acid, and formic acid in a batch reactor at ~150 ℃. It suggests that CO sweeps Rh-CH3 and assists O insertion to form Rh-OCH3 that is further hydrolyzed to methanol. The temperature used in this work is high enough to light off the water gas shift reaction (CO + H2O → H2 + CO2). The other function of CO proposed by the author is to stabilize/regenerate Rh(I) single site [51]. The whole process is pure catalytical, and without using any extra metal (like Cu2+), achieving ~5% methane conversion. More recently, direct use of H2 and O2 to produce H2O2 for methane oxidation has been applied for AuPd/ZSM-5 [50] and Fe/ZSM-5 (combined with Pd catalyst) [47, 48]; however, no significant progress has been made for the activity. A breakthrough was achieved by using a hydrophobic ZSM-5 that encapsulates AuPd NPs (Table 1). The activity boost was ascribed to the fence effect of the hydrophobic molecule that can retain and concentrate local H2O2 for CH4 oxidation [44].

|
Download:
|
| Scheme 1. Proposed reaction mechanism for methane oxidation by CO, H2O, and O2. Reproduced with permission [52]. Copyright 1997, American Chemical Society. | |
|
|
Table 1 Methane oxidation data. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Extensive efforts on catalysts development have been made for methane oxidation. Open supports, such as C for Pd/C [25, 52], TiO2 for Cr/TiO2 [14], Ir/TiO2 [17], and Rh/TiO2 [48] (Rh/ZSM-5 with superior activity) [51], ZrO2 for Rh/ZrO2, SiO2 for Cu/SiO2 [27], C3N4 for Cu/C3N4 [12], Al2O3 for Cu/Al2O3 [53], and SBA-15 for Cu/SBA-15 [54] have been used; Zeolites are extensively studied for different metal based catalysts, such as Cu/zeolites (ZSM-5 [26, 28], SSZ-13 [28], mordenite [26, 28], SAPO-34 [28], and mazzite [55, 56]), Pd/ZSM-5 [15], Pd-Au/ZSM-5 [49, 50], and Fe/ZSM-5 [13]. Even without any support, AuPd alloy can also catalyze methane oxidation to methanol but with extreme poor reactivity in the batch reactor with H2 and O2.
For the most studied catalytic systems, Cu supported by zeolites (ZSM-5 [26, 31], SSZ-13 [31], mordenite [26, 31], and mazzite [55, 56]) show the best performance compared to other non-zeolitic supports. The role of zeolites was proposed to be stabilizing active Cu oxo (Cu-O2-Cu) species; and the zeolites must have the rings that are larger than six members to achieve activity [57-60]. For Pd-Au system, it is even active towards methane partial oxidation without any supports (colloidal) when using H2O2 (O2), and the activity was improved significantly after encapsulating Pd-Au alloy into the zeolites. Since Pd-Au is known for its catalytic activity for H2O2 production, with H2 and O2, the post-silylation of Pd-Au/ZSM-5 (creating hydrophobic fence to concentrate H2O2 in the zeolites) promotes the reaction of methane to methanol substantially [49].
We can intuit that zeolites are the most promising supports for different transition metals containing catalysts (Cu, Pd, and Fe). Using H2, O2, and H2O to partially oxidize methane to methanol is a green method that has potential not only to reduce greenhouse gas emission but also to generate valuable methanol. Moreover, to make the whole process environment friendly, it is desirable to make the catalysts preparation also an environment friendly process.
3. Zeolite synthesis by a green approachAs was suggested above the zeolite support plays a critical role for methane oxidation to methanol, however, zeolite is usually produced by an environmentally unfriendly approach. To make this process more environmentally friendly, it is crucial to generate these zeolite materials in a green way. Synthetic zeolites are usually obtained by hydrothermal crystallization in alkaline media under autogenous pressures in a contained space [61, 62]. Chronological zeolite studies over the decades have informed researchers of the importance of synthetic chemistry development and the selection of organic templates (more commonly referred to as organic structure directing agents, or OSDAs) in successfully crystallizing novel zeolite structures. While the hydrothermal routes have been well-established, there are many aspects that can be innovated to reduce the negative impact on the environment and to improve synthesis process efficiency. From the perspective of sustainable chemistry, the usage of toxic OSDAs should be minimized to limit additional energy consumption and environmental pollution during calcination. The cost of OSDAs is also a concern for large-scale zeolite production since OSDAs are usually more expensive than other raw chemicals and sometimes need to be specially manufactured. To address these challenges, several OSDA-related strategies have been proposed and practiced, such as the usage of low-toxicity templates, low-cost templates, and recyclable templates. On the other hand, the hydrothermal synthesis features insufficient utilization of constituent materials with low zeolite yields and generates abundant aqueous wastes. Thus, novel synthetic routes need to be developed to compliment the established process. In the following sections, an overview of the recent progress on sustainable zeolite synthesis is provided on three topics: (1) OSDA-free synthesis of zeolites, (2) solvent-free synthesis of zeolites and (3) synthesis of 2D zeolites
3.1. OSDA-free synthesis of zeolitesOne notable recent progress on sustainable zeolite synthesis is the efforts to eliminate the usage of organic templates through seed-directed strategies, which can be traced back to the 1960s when the seeds was used to enhance crystallization by eliminating the induction period and to prevent the formation of impurities [63, 64]. While about 30 types of zeolites have been synthesized by such OSDA-free routes [65], most zeolites with unique pore structures or with high silica content still need various OSDAs to template the assembly pathway and to fill the pore spaces. Thus, the development of novel organotemplate-free strategies for zeolite synthesis remains a point of intense investigation in the field of zeolite research. As the concept of green chemistry has received growing attention, the identification of reliable alternative OSDA-free routes has received growing emphasis as a topic of research for zeolite synthesis. One prominent work is the OSDA-free synthesis of beta zeolites when the addition of beta seeds is used to facilitate crystallization of zeolite beta that was once believed to be synthesized only in the presence of organic templates [66]. Due to the success of this case, OSDA-free strategy has been successfully developed to produce many types of zeolites, including PAU, MFI, MEL, FER, MOR, SZR, MSE, MAZ, CHA, RTH, VET, MTW, LEV, EMT, MTT, TON, MWW, and ZSM-34, which represents a critical progress in advancing sustainable chemistry for synthetic zeolites [67]. The OSDA-free zeolites have some unique characteristics on acidity and crystal morphology as compared to the conventional zeolites. First, the OSDA-free zeolites usually have high Al content due to the number of aluminosilicate species needed to balance alkaline cations (e.g., Na+ with high charge densities) [68]. By contrast, the hydrophobic interaction between OSDAs (with low charge density) and silicate species that are prevalent during zeolite synthesis with OSDAs usually leads to high silica zeolites. Second, the mechanistic studies show that the OSDA-free zeolites are grown from the partially dissolved seeds in alkaline precursors and seed growth dominates over secondary nucleation, producing larger zeolite crystals than the seeds, which is a notable morphological feature of the OSDA-free zeolites [69].
For applications, zeolites are required to possess high thermal and chemical stability to be applied as industrial catalysts. Thermally stable zeolites are necessary for catalyst lifetime under reaction conditions, while chemically stable zeolites are needed for any post-synthetic modification to tune acidity and porosity. As the case of OSDA-free beta reveals, while the OSDA-free zeolites exhibit better structural integrity and lower silanol defects with high thermal stability and comparable acid sites strength to the TEA-beta zeolites [70, 71], the chemical stability is still an issue that hinders its usage. The as-synthesized OSDA-free beta zeolite exhibited severe stability issues upon either acidic or basic treatments because of the exceptionally high Al contents [72]. A steaming or static calcination operation is needed to stabilize zeolite framework that a slight dealumination and condensation occurred with the formation of more stable Si‒O‒Si bonds. Without such stabilization, the as-synthesized zeolite can be readily amorphized by exposing them even in diluted acidic or basic solutions. The OSDA-free strategy can be extended to synthesize hierarchical zeolites that exhibit high microporosity and crystallinity due to the occurrence of crystallization under certain hydrothermal conditions [73-75].
3.2. Solvent-free synthesis of zeolitesThe usage of water or other alcohol solvents during zeolite synthesis greatly reduces the yield of zeolite products due to dissolution of silicate/aluminates species while generating polluted wastes. To address these challenges, a solvent-free route was developed for the synthesis of a series of silica, aluminosilicate, and aluminophosphate-based zeolites by simply mixing, grinding, and heating raw materials under solvent-free conditions [76-78]. This route has also been used to synthesize zeolite beta and ZSM-5 without addition of solvents or OSDAs via seed addition [79]. A small yet critical amount of water from the starting materials facilitates the initial hydrolysis of silica species and the subsequent zeolite crystallization. The same route was further optimized to synthesize zeolites MFI, BEA, EUO, and TON from anhydrous silica sources [80]. The synthesized zeolites were shown to possess comparable acidity and catalytic properties to those obtained by conventional hydrothermal synthesis. Up to now, at least twenty types of zeolites have been successfully synthesized by the solvent-free route [81]. It is evident that an effective combination of an OSDA-free approach with a solvent-free route has great potential to maximize the advantages of synthetic process sustainability and efficiency, which minimize the usage of toxic and costly OSDAs while significantly improves zeolite yields through solid-phase transformation.
The solvent-free route also opens opportunities for designing new strategies for metal encapsulation within zeolite channels that aims for synergistically utilizing hydrogenated activities (metal species) and acidity/shape-selectivity (zeolites). Encapsulation of metal and oxide clusters within zeolites can protect them against sintering under reaction conditions and prevent unfavorable contact with toxic impurities, while concurrently allowing active sites to select reactants or transition states based on molecular size within confined zeolite pore channels [82]. For small and medium-pore zeolites, the diffusion limitation or steric hindrance of solvated metal-oxo oligomers poses challenges for effective metal encapsulation within zeolites via post-synthetic protocols. Therefore, there are a series of novel approaches for incorporating metal precursors during synthesis [82-85]. Solvent-free synthesis exhibits great potential for encapsulating metal species into zeolites with highly utilization of metal precursors due to the solid-phase transformation wherein metal nanoparticles can be encapsulated with starting materials (e.g., amorphous silica) before solvent-free crystallization. The resulting core (metal)-shell (zeolite) structured catalysts provided unique performances in terms of selectivity, stability, and versatility [49, 86, 87].
3.3. Synthesis of 2D zeolites2D zeolites refer to zeolite crystalline structures extending in only two dimensions and without structural periodicity in the third dimension. The atoms within the 2D zeolite layers are connected by strong covalent bonds, while the contiguous zeolitic layers are linked by van der Waals forces, hydrogen bonds or ionic interactions. Since the identification of 2D ordered layer precursor (MCM-22P) with MWW topology, different MCM-22P nanosheets assemblies have been generated by a variety of direct and/or post-synthetic modifications, such as calcination, detemplation, swelling, stabilization (interlayer expansion), pillaring, and delamination [88-90]. 2D zeolites can be considered as hosts that can expand and/or extend via structural, topotactic and compositional modifications to form novel and diverse structures. According to the International Zeolite Association (IZA), about 18 zeolite structures have been synthesized as 2D layered structures that can be further extended to over 100 zeolite materials with variations in pore size/shape and chemical composition in zeolitic layers and pillars [91]. Subtle changes in structural and chemical properties are the strong contributors to zeolite catalytic performance via the alteration of acid site strength, location/accessibility, and intrachannel environments.
Among the various modifications, delamination and pillarization have been frequently used to modify 2D zeolites into diverse structures. Delamination separates the stack of 2D zeolite nanosheets into self-standing independent entities by breaking down the interlayer interactions. For example, the delamination of MCM-22P was achieved via a one-step synthesis (MCM-56) [90] or multi-step procedures (ITQ-2, UCB-1) involving pre-swelling, acidification, and/or sonication [92, 93]. Rather than breaking down the stack of 2D zeolites by delamination that may sacrifice structure integrity, the pillarization transforms the 2D zeolites into hierarchical materials with the retention of the stacked layer structure. The current pillaring procedure requires multiple discrete steps, including interlayer expansion by swelling with long chain polar organic molecules, intercalation of the swollen materials in an alkoxide liquid, hydrolysis of entrapped alkoxide in an aqueous alkaline solution, and removal of organics as well as transformation of alkoxide liquid into permanent oxide pillars between zeolitic layers by calcination. Moreover, the pillarization process uses excess solvents in both intercalation and hydrolysis treatments, and generates significant liquid waste, which limit their use in high-scale production operation.
Recently, a vapor-phase pillarization (VPP) process was reported to produce pillared zeolites from 2D layered zeolite structures [94]. The VPP process has ~100% efficiency of alkoxide usage in the intercalation step, requires less (and, in some cases, zero) water addition in the hydrolysis step, does not require separation for product recovery, and generates no liquid waste. Furthermore, synthesis of pillared zeolites via the VPP process can be accomplished within a single apparatus with one-time operation. The pillared 2D ZSM-5 by the VPP process has preserved the structural integrity (both framework crystallinity and ordering of zeolitic nanosheet layers), acidity, and catalytic performance of the zeolite framework in comparison to 2D zeolite prepared using direct calcination. The VPP process is scalable, easy to operate, and highly efficient compared to the previous liquid-phase pillarization method, which could pave a new avenue toward pillaring additional 2D zeolites and new types of layered materials.
4. Concluding remarksThe potential success of catalytic system for methane oxidation to methanol will significantly reduce greenhouse emission during oil production. With the continuing efforts for methane activation to oxygenates, using H2 and O2 in liquid phase stands out as a promising catalytic process. It is essential to have catalytic active sites that can catalyze H2 and O2 to produce H2O2, which will be accessible to methane easily under the reaction conditions. Zeolites play a vital role in achieving high activity and selectivity to methanol product due to their effect in stabilizing active species and concentrating reactant molecules for the active sites. The rational catalyst design that aims the coexistence and intimacy of H2O2 production site and subsequent CH4 activation site has the potential to make this process commercially practical in the future. Moreover, developing green zeolites synthesis approach and new 2D zeolites has the potential to make the methane oxidation to methanol green and efficient.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
| [1] |
A.E. Shilov, G.B. Shul'pin, Russ. Chem. Rev. 56 (1987) 442-464. DOI:10.1070/RC1987v056n05ABEH003282 |
| [2] |
W. Bank, Global Gas Flaring Reduction Partnership, in: W. Bank (Ed. ), https://www.worldbank.org/en/programs/gasflaringreduction.
|
| [3] |
G. Centi, SmartMat 1 (2020) e1005. |
| [4] |
T. Qin, Y. Qian, F. Zhang, B.L. Lin, Chin. Chem. Lett. 30 (2019) 314-318. DOI:10.1016/j.cclet.2018.07.003 |
| [5] |
N. Wang, R.K. Miao, G. Lee, et al., SmartMat 2 (2021) 12-16. DOI:10.1002/smm2.1018 |
| [6] |
S. Wang, S. Hou, C. Wu, Y. Zhao, X. Ma, Chin. Chem. Lett. 30 (2019) 398-402. DOI:10.1016/j.cclet.2018.06.021 |
| [7] |
S.H. Morejudo, R. Zanón, S. Escolástico, et al., Science 353 (2016) 563-566. DOI:10.1126/science.aag0274 |
| [8] |
R.A. Periana, O. Mironov, D. Taube, G. Bhalla, C. Jones, Science 301 (2003) 814-818. DOI:10.1126/science.1086466 |
| [9] |
R.A. Periana, D.J. Taube, S. Gamble, et al., Science 280 (1998) 560-564. DOI:10.1126/science.280.5363.560 |
| [10] |
R.A. Periana, E.R. Evitt, H. Taube, US Patent 5233113, 1993.
|
| [11] |
R.A. Periana, D.J. Taube, H. Taube, E.R. Evitt, US Patent 5306855, 1994.
|
| [12] |
C. Hammond, M.M. Forde, M.H. Ab Rahim, et al., Angew. Chem. Int. Ed. 51 (2012) 5129-5133. DOI:10.1002/anie.201108706 |
| [13] |
W. Huang, S. Zhang, Y. Tang, et al., Angew. Chem. Int. Ed. 55 (2016) 13441-13445. DOI:10.1002/anie.201604708 |
| [14] |
Y. Kwon, T.Y. Kim, G. Kwon, J. Yi, H. Lee, J. Am. Chem. Soc. 139 (2017) 17694-17699. DOI:10.1021/jacs.7b11010 |
| [15] |
Q. Shen, C. Cao, R. Huang, et al., Angew. Chem. 59 (2020) 1216-1219. DOI:10.1002/anie.201913309 |
| [16] |
B. Wu, R. Yang, L. Shi, et al., Chem. Commun. 56 (2020) 14677-14680. DOI:10.1039/D0CC06492K |
| [17] |
J. Xie, R. Jin, A. Li, et al., Nat. Catal. 1 (2018) 889-896. DOI:10.1038/s41929-018-0170-x |
| [18] |
H. Zhou, T. Liu, X. Zhao, et al., Angew. Chem. Int. Ed. 58 (2019) 18388-18393. DOI:10.1002/anie.201912785 |
| [19] |
S.J. Freakley, Q. He, J.H. Harrhy, et al., Science 351 (2016) 965-968. DOI:10.1126/science.aad5705 |
| [20] |
H. Henkel, W. Weber, US Patent 1108752, 1914.
|
| [21] |
R.J. Lewis, G.J. Hutchings, ChemCatChem 11 (2019) 298-308. DOI:10.1002/cctc.201801435 |
| [22] |
Q. Liu, J.C. Bauer, R.E. Schaak, J.H. Lunsford, Appl. Catal. A: Gen. 339 (2008) 130-136. DOI:10.1016/j.apcata.2008.01.026 |
| [23] |
S. Sterchele, P. Biasi, P. Centomo, et al., Appl. Catal. A: Gen. 468 (2013) 160-174. DOI:10.1016/j.apcata.2013.07.057 |
| [24] |
N. Agarwal, S.J. Freakley, R.U. McVicker, et al., Science 358 (2017) 223-227. DOI:10.1126/science.aan6515 |
| [25] |
E.D. Park, Y.S. Hwang, J.S. Lee, Catal. Commun. 2 (2001) 187-190. DOI:10.1016/S1566-7367(01)00030-9 |
| [26] |
M.H. Groothaert, P.J. Smeets, B.F. Sels, P.A. Jacobs, R.A. Schoonheydt, J. Am. Chem. Soc. 127 (2005) 1394-1395. DOI:10.1021/ja047158u |
| [27] |
S.E. Bozbag, P. Sot, M. Nachtegaal, et al., ACS Catal. 8 (2018) 5721-5731. DOI:10.1021/acscatal.8b01021 |
| [28] |
L. Yang, J. Huang, J. Cen, et al., J. Mater. Chem. A 9 (2021) 7094-7101. DOI:10.1039/D0TA10842A |
| [29] |
L. Yang, J. Huang, S. Dai, et al., J. Mater. Chem. A 8 (2020) 24024-24030. DOI:10.1039/D0TA08350J |
| [30] |
L. Yang, J. Huang, R. Ma, et al., ACS Energy Lett. 4 (2019) 2945-2951. DOI:10.1021/acsenergylett.9b01992 |
| [31] |
K. Narsimhan, K. Iyoki, K. Dinh, Y. Román-Leshkov, ACS Cent. Sci. 2 (2016) 424-429. DOI:10.1021/acscentsci.6b00139 |
| [32] |
K.T. Dinh, M.M. Sullivan, K. Narsimhan, et al., J. Am. Chem. Soc. 141 (2019) 11641-11650. DOI:10.1021/jacs.9b04906 |
| [33] |
V.L. Sushkevich, D. Palagin, M. Ranocchiari, J.A. van Bokhoven, Science 356 (2017) 523-527. DOI:10.1126/science.aam9035 |
| [34] |
A. Koishybay, D.F. Shantz, J. Am. Chem. Soc. 142 (2020) 11962-11966. DOI:10.1021/jacs.0c03283 |
| [35] |
Z. Liu, E. Huang, I. Orozco, et al., Science 368 (2020) 513-517. DOI:10.1126/science.aba5005 |
| [36] |
C. Díaz-Urrutia, T. Ott, Science 363 (2019) 1326-1329. DOI:10.1126/science.aav0177 |
| [37] |
R.A. Periana, D.J. Taube, E.R. Evitt, et al., Science 259 (1993) 340-343. DOI:10.1126/science.259.5093.340 |
| [38] |
N. Basickes, T.E. Hogan, A. Sen, J. Am. Chem. Soc. 118 (1996) 13111-13112. DOI:10.1021/ja9632365 |
| [39] |
K. Nakata, Y. Yamaoka, T. Miyata, et al., J. Organomet. Chem. 473 (1994) 329-334. DOI:10.1016/0022-328X(94)80134-7 |
| [40] |
D.G. Piao, K. Inoue, H. Shibasaki, et al., J. Organomet. Chem. 574 (1999) 116-120. DOI:10.1016/S0022-328X(98)00931-0 |
| [41] |
A. Sen, M.A. Benvenuto, M. Lin, A.C. Hutson, N. Basickes, J. Am. Chem. Soc. 116 (1994) 998-1003. DOI:10.1021/ja00082a022 |
| [42] |
N. Takahiro, N. Kazuyuki, T. Ken, F. Yuzo, Chem. Lett. 21 (1992) 1141-1142. DOI:10.1246/cl.1992.1141 |
| [43] |
G. Yin, D.G. Piao, T. Kitamura, Y. Fujiwara, Appl. Organomet. Chem. 14 (2000) 438-442. DOI:10.1002/1099-0739(200008)14:8<438::AID-AOC20>3.0.CO;2-L |
| [44] |
J.K. Edwards, B. Solsona, E. Ntainjua N, et al., Science 323 (2009) 1037-1041. DOI:10.1126/science.1168980 |
| [45] |
P. Xiao, Y. Wang, T. Nishitoba, J.N. Kondo, T. Yokoi, Chem. Commun. 55 (2019) 2896-2899. DOI:10.1039/C8CC10026H |
| [46] |
J. Xu, L. Ouyang, G.J. Da, et al., J. Catal. 285 (2012) 74-82. DOI:10.1016/j.jcat.2011.09.017 |
| [47] |
Z. Jin, L. Wang, E. Zuidema, et al., Science 367 (2020) 193-197. DOI:10.1126/science.aaw1108 |
| [48] |
J. Kang, E.D. Park, Catalysts 10 (2020) 299. DOI:10.3390/catal10030299 |
| [49] |
J. Kang, P. Puthiaraj, W.S. Ahn, E.D. Park, Appl. Catal. A: Gen. 602 (2020) 117711. DOI:10.1016/j.apcata.2020.117711 |
| [50] |
R.J. Lewis, A. Bara-Estaun, N. Agarwal, et al., Catal. Lett. 149 (2019) 3066-3075. DOI:10.1007/s10562-019-02876-7 |
| [51] |
J.J. Shan, M.W. Li, L.F. Allard, S. Lee, M. Flytzani-Stephanopoulos, Nature 551 (2017) 605-608. DOI:10.1038/nature24640 |
| [52] |
M. Lin, T. Hogan, A. Sen, J. Am. Chem. Soc. 119 (1997) 6048-6053. DOI:10.1021/ja964371k |
| [53] |
J. Meyet, K. Searles, M.A. Newton, et al., Angew. Chem. Int. Ed. 58 (2019) 9841-9845. DOI:10.1002/anie.201903802 |
| [54] |
H.V. Le, S. Parishan, A. Sagaltchik, et al., Chem. Eur. J. 24 (2018) 12592-12599. DOI:10.1002/chem.201801135 |
| [55] |
A.J. Knorpp, A.B. Pinar, M.A. Newton, V.L. Sushkevich, J.A. van Bokhoven, ChemCatChem 10 (2018) 5593-5596. DOI:10.1002/cctc.201801809 |
| [56] |
M.B. Park, S.H. Ahn, A. Mansouri, M. Ranocchiari, J.A. van Bokhoven, ChemCatChem 9 (2017) 3705-3713. DOI:10.1002/cctc.201700768 |
| [57] |
D.W. Fickel, R.F. Lobo, J. Phys. Chem. C 114 (2010) 1633-1640. |
| [58] |
A. Godiksen, F.N. Stappen, P.N.R. Vennestrøm, et al., J. Phys. Chem. C 118 (2014) 23126-23138. DOI:10.1021/jp5065616 |
| [59] |
D.K. Pappas, E. Borfecchia, M. Dyballa, et al., J. Am. Chem. Soc. 139 (2017) 14961-14975. DOI:10.1021/jacs.7b06472 |
| [60] |
P.J. Smeets, R.G. Hadt, J.S. Woertink, et al., J. Am. Chem. Soc. 132 (2010) 14736-14738. DOI:10.1021/ja106283u |
| [61] |
C.S. Cundy, P.A. Cox, Chem. Rev. 103 (2003) 663-702. DOI:10.1021/cr020060i |
| [62] |
Y. Xie, W. Yuan, Y. Huang, et al., Chin. Chem. Lett. 30 (2019) 359-362. DOI:10.1016/j.cclet.2018.03.026 |
| [63] |
G.T. Kerr, J. Phys. Chem. 70 (1966) 1047-1050. DOI:10.1021/j100876a015 |
| [64] |
G.T. Kerr, J. Phys. Chem. 72 (1968) 1385-1386. DOI:10.1021/j100850a056 |
| [65] |
M.D. Oleksiak, J.D. Rimer, Rev. Chem. Eng. 30 (2014) 1-49. DOI:10.1515/revce-2013-0020 |
| [66] |
B. Xie, J. Song, L. Ren, et al., Chem. Mater. 20 (2008) 4533-4535. DOI:10.1021/cm801167e |
| [67] |
K. Zhang, S. Fernandez, M.L. Ostraat, ChemCatChem 10 (2018) 4197-4212. DOI:10.1002/cctc.201800612 |
| [68] |
K. Iyoki, K. Itabashi, T. Okubo, Micropor. Mesopor. Mat. 189 (2014) 22-30. DOI:10.1016/j.micromeso.2013.08.008 |
| [69] |
Y. Kamimura, S. Tanahashi, K. Itabashi, et al., J. Phys. Chem. C 115 (2011) 744-750. DOI:10.1021/jp1098975 |
| [70] |
B. Yilmaz, U. Müller, M. Feyen, et al., Catal. Sci. Technol. 3 (2013) 2580-2586. DOI:10.1039/c3cy00073g |
| [71] |
H. Zhang, B. Xie, X. Meng, et al., Micropor. Mesopor. Mat. 180 (2013) 123-129. DOI:10.1016/j.micromeso.2013.06.031 |
| [72] |
K. Zhang, S. Fernandez, J.A. Lawrence, M.L. Ostraat, ACS Omega 3 (2018) 18935-18942. DOI:10.1021/acsomega.8b02762 |
| [73] |
S. Fernandez, M.L. Ostraat, K. Zhang, AIChE J. 66 (2020) e16943. |
| [74] |
K. Zhang, S. Fernandez, E.S. Converse, S. Kobaslija, Catal. Sci. Technol. 10 (2020) 4602-4611. DOI:10.1039/D0CY01209B |
| [75] |
K. Zhang, S. Fernandez, J.T. O'Brien, et al., Catal. Today 316 (2018) 26-30. DOI:10.1016/j.cattod.2017.11.033 |
| [76] |
Y. Jin, X. Chen, Q. Sun, et al., Chem. Eur. J. 20 (2014) 17616-17623. DOI:10.1002/chem.201403890 |
| [77] |
Y. Jin, Q. Sun, G. Qi, et al., Angew. Chem. 52 (2013) 9172-9175. DOI:10.1002/anie.201302672 |
| [78] |
L. Ren, Q. Wu, C. Yang, et al., J. Am. Chem. Soc. 134 (2012) 15173-15176. DOI:10.1021/ja3044954 |
| [79] |
Q. Wu, X. Wang, G. Qi, et al., J. Am. Chem. Soc. 136 (2014) 4019-4025. DOI:10.1021/ja500098j |
| [80] |
Q. Wu, X. Liu, L. Zhu, et al., J. Am. Chem. Soc. 137 (2015) 1052-1055. DOI:10.1021/ja5124013 |
| [81] |
Q. Wu, X. Meng, X. Gao, F.S. Xiao, Acc. Chem. Res. 51 (2018) 1396-1403. DOI:10.1021/acs.accounts.8b00057 |
| [82] |
S. Goel, S.I. Zones, E. Iglesia, J. Am. Chem. Soc. 136 (2014) 15280-15290. DOI:10.1021/ja507956m |
| [83] |
M. Choi, Z. Wu, E. Iglesia, J. Am. Chem. Soc. 132 (2010) 9129-9137. DOI:10.1021/ja102778e |
| [84] |
S. Goel, Z. Wu, S.I. Zones, E. Iglesia, J. Am. Chem. Soc. 134 (2012) 17688-17695. DOI:10.1021/ja307370z |
| [85] |
Z. Wu, S. Goel, M. Choi, E. Iglesia, J. Catal. 311 (2014) 458-468. DOI:10.1016/j.jcat.2013.12.021 |
| [86] |
C. Wang, L. Wang, J. Zhang, et al., J. Am. Chem. Soc. 138 (2016) 7880-7883. DOI:10.1021/jacs.6b04951 |
| [87] |
L. Wang, G. Wang, J. Zhang, et al., Nat. Commun. 8 (2017) 1-8. DOI:10.1038/s41467-016-0009-6 |
| [88] |
U. Díaz, A. Corma, Dalton Trans. 43 (2014) 10292-10316. DOI:10.1039/c3dt53181c |
| [89] |
W.J. Roth, P. Nachtigall, R.E. Morris, J. Cejka, Chem. Rev. 114 (2014) 4807-4837. DOI:10.1021/cr400600f |
| [90] |
K. Zhang, M.L. Ostraat, Catal. Today 264 (2016) 3-15. DOI:10.1016/j.cattod.2015.08.012 |
| [91] |
E. Schulman, W. Wu, D. Liu, Materials 13 (2020) 1822. DOI:10.3390/ma13081822 |
| [92] |
A. Corma, V. Fornes, S. Pergher, T.L. Maesen, J. Buglass, Nature 396 (1998) 353-356. DOI:10.1038/24592 |
| [93] |
I. Ogino, M.M. Nigra, S.J. Hwang, et al., J. Am. Chem. Soc. 133 (2011) 3288-3291. DOI:10.1021/ja111147z |
| [94] |
L. Wei, K. Song, W. Wu, et al., J. Am. Chem. Soc. 141 (2019) 8712-8716. DOI:10.1021/jacs.9b03479 |