 2022, Vol. 33
2022, Vol. 33 


b Beijing National Laboratory for Molecular Sciences (BNLMS), State Key Laboratory for Structural Chemistry of Unstable and Stable Species, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
Group IB metal nanoparticles have been successfully used as oxidation catalysts in ethylene epoxidation [1], partial oxidation of methanol [2], complete oxidation of formaldehyde [3], and other chemical reactions [4]. Meanwhile, copper oxides are used as pigments, p-type semiconductors, as well as dry cell batteries [5, 6]. Joint experimental and theoretical studies have revealed the diverse structures and properties of oxo, peroxo and superoxo isomers of several neutral and charged species CuOn (n = 1−6) and Cu2On (n = 1−4) [7-10]. On-going efforts are also devoted to exploring the gas-phase reactivity of copper cluster with O2 [11-14]. For example, Ichihashi and coworkers [13] studied the reactions of size-selected copper cluster ions under single collision conditions, and found the reactive cross section of cationic Cun+ was governed by the electronic shell structure, whereas the anionic ones did not exhibit significant variation whether or not electronic shell closing. Among others, reasonable research interest has been stimulated to understand the basic chemical properties of novel copper oxides [15, 16], to attain catalytic activation of molecular oxygen in mild conditions [17, 18], and to unveil the structure-activity relationships of copper-related materials at reduced sizes [19, 20].
Previous studies have illustrated the interactions between O2 and the coinage metal cluster anions, and two reaction mechanisms have been proposed for the reaction between O2 and silver (or gold) anionic clusters: (ⅰ) electron transfer; (ⅱ) spin accommodation. The key insight in electron transfer model is that chemisorption interaction between Ag2n− (or Au2n−) clusters with O2 changes from Ag2n−·O2 to a lower energy adsorbed state Ag2n·O2− [21, 22]. For example, Xing et al. [22, 23] demonstrated that oxygen can be activated only when the adiabatic ionization energy (ADE) of cluster is lower than a certain threshold (3.0 or 3.5 eV for Ag2n− and Au2n− respectively), which can be explained by a scheme that a lower ADE of Ag2n− corresponds to a reasonable preparation energy in forming Ag2n−·O2, and results in a larger binding energy to attain the Ag2n·O2− state pertaining to electron transfer. This model is helpful to predict the reaction trend of even-sized coinage metal clusters from one aspect [18, 21, 23-26].
In view of the unique electron configuration (n−1)d10ns1, coinage metal clusters bear alternative open/closed electronic shell and readily exhibit odd-even oscillation of highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) gaps and binding energies. In particular, when reacting with ground state triplet oxygen (3O2), the odd-even oscillation will be associated with spin accommodation [27-29]. A successful example has been illustrated for typical silver cluster anions [27]. According to the Wigner-Witmer rules of spin conservation [30, 31], spin-orbital effects of Ag and O atoms can be negligible. On this basis, the spin of an unpaired electron on Ag2n− cluster can be aligned opposite to that of 3O2 molecule and simply transfer to 3O2 molecule, which finally results in 2Ag2n·O2− products. Whereas, for the odd-numbered Ag2n+1− clusters, spin crossing occurs in order to occupy the half-filled antibonding orbitals of 3O2 molecule. The reaction process is written as [27]:

|
(1) |
The novel spin accommodation involved in the reaction process for odd-number silver clusters is evidenced by the observation of prominent stability of Ag13−, which bears larger spin excitation energy than its neighboring counterparts [27].
In this work, by employing the laser evaporation (LaVa) metal cluster source, we have prepared a series of copper cluster anions Cun– (n = 7−41) and monitored their reactions with O2 molecules. By combining ab initio calculations of Cun– (n = 7−20) clusters, the structure-activity relationship between Cun– and O2 as well as the decomposition mechanism of O2 on Cun– clusters are systematically investigated. We find the weaker activity of the Cun– clusters toward O2 is associated with the lower energy of the HOMO and the higher vertical detachment energy (VDE). While the spin accommodation does play a determining role in initiating the copper cluster reacting with oxygen, we propose that the increase of edge and hollow sites provides convenience for oxygen bonding and enables leveling effect of the odd-even alternating reactivity.
The experimental instrument composed of a laser ablation source, a fast-flow reactor, and a high resolution of reflection time-of-flight mass spectrometer (Re-TOFMS). The Cun– (n = 7‒41) anions were generated by laser ablation of a copper disk (99.99% pure, 16 mm diameter, 3 mm thickness) and helium (99.999% purity) as a carrier gas with a backing pressure of 10 standard atmospheres. The generated clusters were expanded and reacted with oxygen in a fast-flow reactor (length ~60 mm) where the pressure of carrier gas was about 55 Pa at T = 298 K. The effective reaction time in the reactor was estimated to be 60 µs. The reactants and product ions passed through a skimmer (2 mm diameter) at the end of the fast flow tube reactor and were directed to the Re-TOFMS to be analyzed. Other details of the experimental instrument can be found in the previous publications by Luo's group [32].
The comprehensive genetic algorithm (CGA) code developed by Zhao's group [33, 34] was employed to unbiased global search the potential energy surfaces of Cun– (n = 7−20) clusters, accompanied with density functional theory (DFT) calculations operated on DMol3 package [35]. Totally 5000 iterations were lasted to ensure the global minimum on the potential energy surface for Cun– clusters. All structures obtained by CGA search were optimized using the double numerical plus polarization d-function basis (DND) and the GGA-PBE [36] functional under all-electron relativistic method. Sixteen low-energy isomers from CGA-DFT were re-optimized to obtain the lowest-energy structures under the HSE06 [37] functional using Gaussian16 software [38]. The SDD [39] and 6-311+G(d) [40] basis sets were selected for Cu and O atoms, respectively. Moreover, energy decomposition analysis and natural orbitals for chemical valence (EDA-NOCV) [41] method were employed to investigate the interaction and bonding nature between cluster and O2 molecule by performing single point energy calculations using ADF2018.104 [42, 43]. The all-electron Slater basis set of triple-zeta with polarization function (TZP) were selected for calculation and the scalar-relativistic effect was considered under the zero-order regular approximation (ZORA).
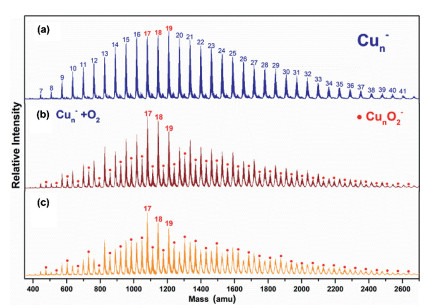
Fig. 1a presents a typical TOF mass spectrum of the Cun– (n = 7−41) clusters, showing a normal Gaussian distribution on the mass abundance. By introducing O2 gas into the fast-flow reactor, the normal distribution broke down as the new peak signal intensity of CunO2− clusters anion increased (Figs. 1b and c). It is expected that the following O2 adsorption reaction (2) proceeds:
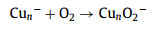
|
(2) |

|
Download:
|
| Fig. 1. Typical TOF mass spectra of copper cluster anions produced via (a) the laser ablation source and after exposure to different partial pressure of O2 gas at (b) 39 mPa and (c) 62 mPa. The numbers of atoms in copper cluster anions are labeled on top of panel (a). The products CunO2– are labeled as black solid circles (●). | |
On this basis, the pseudo-first-order rate constants (k1) for the reactions between Cun− (n = 7−41) and O2 in the fast reactor can be estimated using Eq. 3 below, where the IA and I are the integrated area of mass spectral peaks of copper clusters before and after the reaction, respectively.
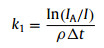
|
(3) |
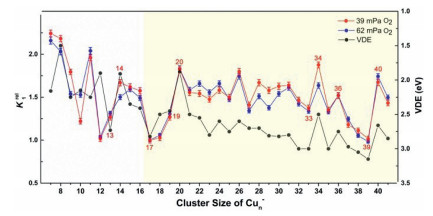
Here ρ is the molecular density of the reactant gas and Δt is the effective reaction time in the reactor (~60 µs). A size-dependent reactivity has been identified through the relative rate constants k1rel = k1(Cun– + O2)/k1(Cu17– + O2) for the reactions between Cun– (n = 7−41) and O2 (Fig. 2). This is consistent with our initial studies on this topic by reacting Cun– with O2 under a smaller gas pressure (~32 mPa) [44], where the same trend of reaction rate was obtained. It is worth mentioning that, while we tried to collect the similar distribution of the copper clusters in probing their reactivities with different chemicals, it is challenging to control exactly the same concentrations of the clusters and the different reactants. This is partly because the copper cluster anions are rather reactive even in presence of trace amount of contamination, and the Cu−Cu bond energy is relatively small (201 kJ/mol). Notably, the reaction rate constants are very well consistent with the VDE values as estimated in the previous study [45]. Also, this is generally consistent with the results in Ichihashi's report [13], where the dominant reaction products Cun-2O2– and Cun-1O2– were observed for n = 3−16 and CunO2– was detected as the main product in n ≥ 17 at the collision energy of 0.2 eV under the single collision condition.

|
Download:
|
| Fig. 2. A comparison of the experimentally determined relative rate constants k1rel = k1(Cun− + O2) /k1(Cu17− + O2) for the reactions between Cun− (n = 7−41) and O2, with the data of VDEs of the Cun− clusters from reference [45]. | |
As shown in Fig. S1 (Supporting information), the stacking motif of Cun− clusters changes from capped octahedron (Cu7−9−) to oblate architecture (Cu10−15−) and finally to core-shell configuration (Cu16−20−). At Cu17−, the cluster has a high symmetrical caged Td structure. Accompanied with all the triangle surfaces of the cluster, this cluster can be assigned into a Frank-Kasper polyhedron. The Cu18− cluster also has a high symmetrical geometry which can be viewed as two Cu atoms capped on the opposite rectangle faces of Cu@Cu15 cage. Cu17− and Cu18− clusters are doubly closed and open shell superatoms with 1S21P61D10 and 1S21P61D102S1 electronic configuration (Fig. S2 in Supporting information), respectively. More details can be found in our previous papers [44, 46].
The energetic properties of Cun− (n = 7−20) clusters are presented in Table 1, including incremental binding energies (IBE), second-order difference in binding energy (∆2E), highest occupied molecular orbital-lowest unoccupied molecular orbital (H-L) gap, vertical spin excitation energies (VSE), and VDEs. Specifically, IBE and ∆2E are defined as:
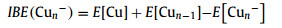
|
(4) |
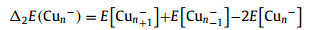
|
(5) |
|
|
Table 1 Electronic properties of Cun− clusters as presented in Fig. S1, including minimum vibration frequencies (ν, in cm−1), incremental binding energies (IBE, in eV), the second-order difference in binding energies (∆2E, in eV), average bond lengths between copper atoms (R, in Å), highest occupied molecular orbital-lowest unoccupied molecular orbital gaps (H-L, in eV) from energy levels of alpha states, vertical spin excitation energies (VSE, in eV), and theoretical and experimental [45] vertical detachment energies (VDEt, VDEe, in eV). |
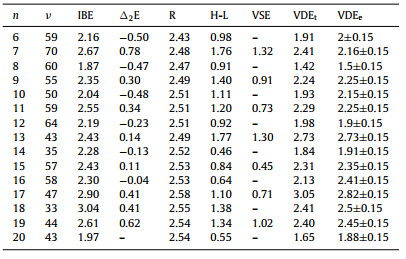
Generally speaking, IBE, ∆2E, H-L gap, and VDE all exhibit odd-even oscillation (except for n = 18), indicating that the clusters with open shell are more active. Among all cluster sizes, Cu18− cluster has the largest IBE of 3.04 eV, followed by 2.90 eV for Cu17−. Besides, Cu18− has the largest ∆2E (0.41 eV) and H-L gap (1.38 eV) among all even-sized clusters. For the VDEs of clusters, the Cu17− cluster has the largest value of 3.05 eV, and Cu13−, Cu18−, and Cu19− clusters show appreciable values of 2.73 eV, 2.41 eV and 2.40 eV, respectively. All energetic results indicate the remarkable stability of Cu17− and Cu18− clusters, which can be related to their superatomic character. In the following, we will discuss O2 molecular adsorption behavior on Cun− clusters in detail and unveil the main energetic factors that dominate the cluster activity.
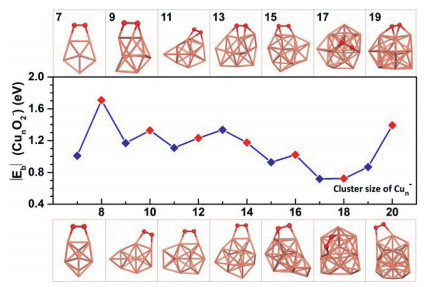
After screening large amounts of active sites for each size of Cun− (n = 7−20) cluster, we obtained the lowest-energy geometries (coordinates in Table S2 in Supporting information) of O2 molecule adsorbed, as depicted in Fig. 3. The O2 molecule is favored to locate on the bridge sites of even-sized clusters (except for Cu16−), while the O2 molecule prefers the hollow sites of odd-sized clusters (except for Cu7−). The Cu2nO2− and Cu2n+1O2− complexes are in doublet and singlet spin states (except for Cu7O2− being triplet), respectively. To evaluate the activity of Cun− cluster toward O2, we calculated the adsorption energies (Eb) of O2 chemisorbed on clusters using the following formula:
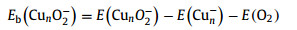
|
(6) |

|
Download:
|
| Fig. 3. Structures of molecular oxygen adsorbed on Cun− (n = 7−20) clusters. The complexes are in singlet and doublet spin states for clusters with odd and even number of copper atoms (except for Cu7O2− in triplet state), respectively. | |
The Cun− (n = 7−20) clusters show strong activities toward O2 with adsorption energies are all lower than −0.7 eV, thereby inducing the intensive geometrical reconstruction of clusters during the process of chemisorption of O2 [22, 46]. Overall, the adsorption energies of O2 molecule on Cun− (n = 7−20) clusters in Fig. 3 exhibit odd-even oscillation with two interrupted points at the cluster sizes of n = 13 and 18. The adsorption energies of O2 on Cun− (n = 7−20) clusters fall in the range similar to those previous theoretical reports on Cu(111) and Cu(110) surfaces [47]. And the trend of O2 adsorption energies on Cun− (n = 14−20) clusters is consistent with the experimental relative rate constants in Fig. 2. Natural population analysis (NPA) charge in Fig. S3 (Supporting information) discloses that the odd- and even-sized copper cluster anions transfer 1.18−1.35|e| and 0.64−0.69|e| to O2, respectively. Correspondingly, the odd-sized clusters can elongate the O−O bond by 0.26−0.30 Å, while the even-sized clusters can only slightly elongate the O−O bond by about 0.13 Å.
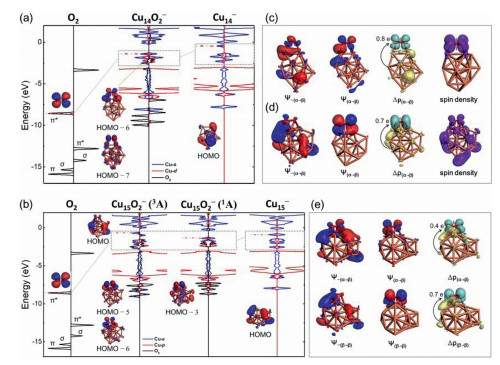
Herein the Cu14− and Cu15− clusters were selected as representatives, considering a balance of calculation efficiency and calculation accuracy, and the two typical clusters both follow the oxidation law as most of the other copper clusters. We presented the total and partial density of states (PDOS) of O2, pure cluster, and 2Cu14O2−, 3Cu15O2−, 1Cu15O2− complexes in Fig. 4. From the PDOS of complexes, the π* and σ orbitals of O2 molecule strongly hybridize with frontier orbitals and energy levels under the d-band of cluster, respectively. As for 2Cu14O2− complex, π* orbitals of O2 are hybrid with frontier orbitals of the cluster to form the HOMO−6 and HOMO−7 levels. In view of 3Cu15O2−, the orbital hybridization between O2 and cluster does not stabilize the HOMO energy level of complex; in addition, it also reduces the gap. Contrarily, 1Cu15O2− in singlet state lowers the energy level of HOMO, increases the HOMO-LUMO gap in comparison with the pristine cluster, suggesting a higher chemical inertness.

|
Download:
|
| Fig. 4. (a) From left to right: DOS (or PDOS) of the free O2 molecule, O2 chemisorbed on the Cu14− cluster, and the pristine Cu14− cluster, respectively. (b) From left to right: DOS (or PDOS) of the free O2 molecule, O2 chemisorbed on the Cu15− cluster in triplet (at the same structure as singlet state) and singlet state, and the pristine Cu15− cluster, respectively. NOCV pairwise of orbitals Ψ−n/Ψn, and the corresponding deformation electron density ∆ρn between (c) O2 and Cu14− fragments, (d) O2 and Cu15− fragments of triplet 3Cu15O2− complex, and (e) O2 and Cu15− fragments of singlet 1Cu15O2− complex. The isosurface values for electron cloud distribution and deformation electron density are ± 0.04 a.u. and ± 0.0015 a.u., respectively. The cyan and yellow colors of ∆ρn represent electron accumulation and depletion regions, respectively. The electrons transferred from clusters to O2 molecule analyzed by NOCV method are showed in ∆ρn illustrations. | |
Furthermore, we selected the clusters in their lowest-energy spin multiplicity and triplet O2 as two fragments to analyze their chemical bonding using EDA-NOCV method and further clarified the mechanism of O2 adsorption on Cun− (n = 7−20) clusters. In this method, the total bonding energy of system is divided into three parts, including repulsion energy caused by the Pauli exclusion (ΔEpauli), attractive energies from electrostatic (ΔEelstat), and orbital interactions (ΔEoi):
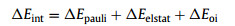
|
(7) |
As shown in Table S1 (Supporting information), the stabilities of these complexes come from the orbital interactions by 53−57 percent and else from the electrostatic interactions between two fragments. The electron cloud shape of the pairwise orbital interaction between two fragments before (ψ−n) and after (ψn) chemical bonding as well as electron deformation density plots of pairwise orbital interactions of doublet Cu14O2−, triplet and singlet Cu15O2− complexes are presented in Figs. 4c-e (the results of other sized complexes are given in Table S1). As shown in Fig. 4c, the pairwise NOCV orbital interaction reveals that activation process involving the α−HOMO energy level of Cu14− donates about one electron to the β−π* orbital of O2 between two fragments, which is labeled as α→β thereafter. After that, there are no lone pair electrons on Cu14− cluster, while O2 molecule has a lone-pair electron on its π* orbitals (see spin density in Fig. 4c). The HOMO−7 contributes to the magnetic moment of the complex, which can be proved by comparing the electron cloud shape of the energy level (Fig. 4a) and spin density (Fig. 4c) of system.
Similar to Cu14O2−, the α→β orbital interaction is also observed between Cu15− cluster and O2 in the triplet state of 3Cu15O2−. After losing one electron, the HOMO of cluster as well as π* orbitals of O2 retain a lone-pair electron, which endows the whole complex triplet state, as shown in Fig. 4d. The mülliken analysis indicates that the magnetic moments of 3Cu15O2− are 0.39 μB and 1.61 μB from the π* orbitals of O2 molecule and the HOMO of Cu15− cluster, respectively (see Figs. 4b and d for details). Surprisingly, in the case of singlet 1Cu15O2− complex, not only the α→β orbital interaction, but also another pairwise NOCV orbital interaction is found, that is, β−HOMO of cluster donates about one electron to the π* orbitals of O2 (marked as β→β thereafter). Such additional β→β orbital interactions in Cu2n+1O2− complexes significantly increase the orbital interaction energies for Cu2n+1O2− complexes, making the total bonding energies of Cu2n+1O2− complexes lower than those from Cu2nO2− complexes (Table S1); meanwhile O2 in Cu2n+1O2− complexes are more thoroughly activated than that in Cu2nO2− complexes. This leads to the NPA charges of O2 on Cu2n+1O2− about twice as large as those on Cu2nO2− (Fig. S3) and the weaker O−O bond on Cu2n+1O2− (bond length 1.45−1.49 Å) than those on Cu2nO2− (bond length 1.31−1.32 Å). Moreover, these α→β and β→β interactions in Cu2n+1O2− complexes indicate both π* orbitals of O2 are occupied, which results in the spin transition of O2 from triplet in the gas phase to an adsorbed state with a zero net spin [48]. As discussed above, it can be proved from both geometric and electronic points of view that the superoxo-like and peroxo-like binding of O2 occur on Cu2n− and Cu2n+1− clusters, respectively [25, 48]. According to this scheme, we propose that the adsorption of oxygen on Cu2n− and Cu2n+1− clusters follows single and double electron transfer model, respectively.
Both of these two models indicate that the activity of anionic copper cluster toward O2 is sensitively related to the energy of HOMO (or the VDE value). Besides, the activity of odd-sized clusters also depends on its VSE (or the adiabatic spin excitation energy (ASE)) because of the demand for the conservation of total spin. Keeping this in mind, we have drawn the diagram of VDEs and HOMOs for Cun− (n = 7−42) and Cun− (n = 7−20) clusters in Fig. 2 and Fig. S4 (Supporting information), respectively, and further compared their variation with cluster size with the reaction rate diagram. One can see that, the lower of the HOMO level, the larger VDE value, and the slower the reaction rate. The oscillation of relative rate constants is generally correlated with VDE values of Cun− reported in literature, implying that the ability to donate electrons of anionic copper clusters plays a crucial role in activating oxygen molecule. Additionally, we find that the odd-sized Cu15− cluster with smallest VDE as well as smallest VSE (also for ASE) react fastest with O2, while Cu17− with the largest VDE or Cu19− cluster with largest VSE (also for ASE) reacts with O2 slower than Cu15− cluster (details in Fig. S5 in Supporting information). All the above analyses support the rationality of our explanation of adsorption mechanism.
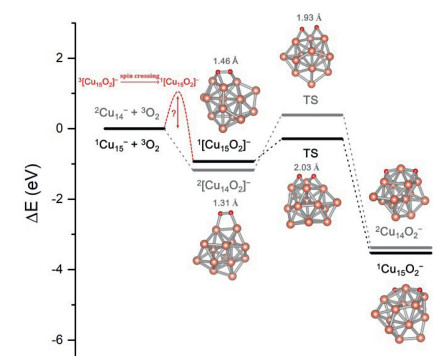
The dissociation of O−O bond of molecular O2 yielding two O atoms adsorbed on the metal surface is usually considered as the first fundamental step in the metal-catalyzed oxidation reactions [47]. Here we systematically investigated this elementary step over the reaction O2 molecule on various sizes of the Cun− clusters. Typically, a comparison of "2Cu14− + 3O2" and "1Cu15− + 3O2" is given in Fig. 5. As demonstrated by the DFT calculations, the formations of 2Cu14O2− and 1Cu15O2− are both highly exothermic (up to ~4 eV), and the two reaction paths are actually comparable in view of the fact that the former needs to overcome spin crossing while the latter has a relatively higher energy barrier for the transition state. Similar results of the other systems have also been obtained (see details in Table 2). According to the ability of accepting and donating electrons, here we make analogy of oxygen and clusters to acids and bases, respectively. Following the above discussion, the Cu2n+1O2− clusters show stronger basicity than Cu2nO2− clusters since they can donate twice as much electron or charge to oxygen as the open shell clusters. This strong acid-base interaction between Cu2n+1− clusters and O2 counteracts the spin transition barrier for the singlet state product from 3Cu2n+1O2− complexes. Such phenomenon of metal clusters with different basicity to activate oxygen by adjusting charge transfer along with spin accommodation is, to some extent, analogous to the leveling effect in acid-alkali chemistry due to autoprotolysis of solvent [49].

|
Download:
|
| Fig. 5. Energy and corresponding atomic structure of reaction intermediates for the reactions between Cu14, 15− clusters and O2. | |
|
|
Table 2 Adsorption energies of O2 on the Cun− clusters (Eb), activation (Eact) and reaction (∆E) energies, calculated at HSE06/SDD/Cu/6-311+G(d)/O level of theory, together with the O−O distances (RO-O) in the TSs. The corresponding geometries of the reaction intermediates are depicted in Figs. 3, 5 and 6. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
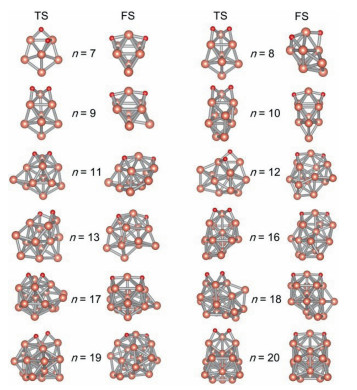
As shown in Fig. 6, the transition state (TS) for the dissociation of the O−O bond and the final state (FS) have been optimized (coordinates are in Tables S3 and S4 in Supporting information). TSs and kinetic barriers for dissociation of O2 were determined by Berny algorithm [50]. According to the observation of TSs, during the rupturing process of O−O bond (elongating to 1.91−2.23 Å in Table 2); the stability of the complex was enhanced by adding at least one Cu−O bond. The positions of two O atoms on Cun− clusters in TSs can be divided into three types: (ⅰ) both two O atoms locating at the bridge conformation, (ⅱ) both two O atoms locating at the hollow conformation, (ⅲ) one O atom and another locating at the bridge site and hollow site, respectively. The clusters with these three types of O−O structures occurred for Cun− (n = 7−10, 12, 14−20), Cu11− and Cu13−, respectively. For the geometries of final products, the O−O bond is completely fractured; O atoms are located on the opposite faces of clusters, sharing a Cu atom in the middle. Such O−O conformations in TSs and FSs have also been observed in previous work [12].

|
Download:
|
| Fig. 6. Structures of TS and FS for the process of dissociation of O2 molecule on Cun− (n = 7−13, 16−20) clusters. | |
{kind=link}
Further, the activation energy (Eact) and reaction energy (ΔE) were calculated to discuss the dissociation process of O2 on Cun− clusters, which are defined by:
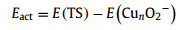
|
(8) |

|
(9) |
Compared with previous studies, the negatively charged Cun− are more active than their neutral [12] and cationic [13] counterparts. However, the energy released by O2 adsorption on these clusters is too small to be used to disrupt the O−O bond; thus, the complex needs additional energy to complete the dissociation process. As shown in Table 2, the Cu7−13− clusters exhibit relatively large negative values of Eb, while relatively small Eact values, indicating feasible O−O bond dissociation. While there are also large adsorption energies for O2 on Cu14− (−1.18 eV) and Cu20− (−1.39 eV) clusters, the large activation energies up to 1.57 and 1.66 eV make them less active. Among them, the Cu8−10, 12−13− clusters are more advantageous for the dissociation of O2 than copper surfaces Cu(111), Cu(110), Cu(100), which always requires extra energies [47]. For the other clusters, the required energy in the dissociation process is between 0.06 eV and 0.27 eV, which is very close to the reported data of 0.03−0.17 eV for oxygen on the copper surface [47].
In summary, the adsorption and dissociation of O2 molecule on Cun− (n = 7−20) cluster anions are systemically investigated by combining the experimental mass spectrometry and ab initio calculations. We found that both open and closed shell Cun− clusters can react with O2 and the overall reaction rates show odd-even oscillation. Such unique activity of closed shell Cun− clusters brings us a new interaction mechanism: double electron transfer. Accompanied with the single electron transfer from open shell Cun− clusters to O2, we proposed the "leveling effect" of spin accommodation for the phenomenon of clusters with different basicity to activate oxygen. Pairwise NOCV orbital interactions indicate that, the lower HOMO, the larger VDE, and the weaker activity of cluster. Besides, the activities of odd-sized clusters also depend on their VSE (or ASE) owing to the spin excitation occurred in the O2 chemisorb process. During the dissociation process of O2, the O−O bond will firstly elongate to 1.91−2.23 Å and two O atoms are located at bridge sites of the Cun− clusters (rate-limiting steps) and finally capped on two opposite faces of the clusters.
Declaration of competing interestWe declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 91961204, 11974068, 21802146, 21722308), the Fundamental Research Funds for the Central Universities of China (No. DUT20LAB110). The authors acknowledge the computer resources provided by the Shanghai Supercomputer Center.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.08.127.
| [1] |
A.J.F. van Hoof, I.A.W. Filot, H. Friedrich, E.J.M. Hensen, ACS Catal. 8 (2018) 11794-11800. DOI:10.1021/acscatal.8b03331 |
| [2] |
A.N. Pestryakov, V.V. Lunin, N. Bogdanchikova, O.N. Temkin, E. Smolentseva, Fuel 110 (2013) 48-53. DOI:10.1016/j.fuel.2012.10.012 |
| [3] |
Z. Huang, X. Gu, Q. Cao, et al., Angew. Chem. Int. Ed. 51 (2012) 4198-4203. DOI:10.1002/anie.201109065 |
| [4] |
J. Teržan, M. Huš, B. Likozar, P. Djinović, ACS Catal. 10 (2020) 13415-13436. DOI:10.1021/acscatal.0c03340 |
| [5] |
W.J. Qi, C.Z. Huang, L.Q. Chen, Talanta 80 (2010) 1400-1405. DOI:10.1016/j.talanta.2009.09.042 |
| [6] |
X.L. Xu, B. Yang, Z.Y. Wei, et al., Phys. Chem. Chem. Phys. 20 (2018) 20622-20628. DOI:10.1039/C8CP03302A |
| [7] |
Y. Gong, G. Wang, M. Zhou, J. Phys. Chem. A 113 (2009) 5355-5359. DOI:10.1021/jp902166x |
| [8] |
H. Wu, S.R. Desai, L.S. Wang, J. Phys. Chem. A 101 (1997) 2103-2111. DOI:10.1021/jp9631442 |
| [9] |
K. Deng, J. Yang, Q. Zhu, J. Chem. Phys. 113 (2000) 7867-7873. DOI:10.1063/1.1316043 |
| [10] |
C. Massobrio, Y. Pouillon, J. Chem. Phys. 119 (2003) 8305-8310. DOI:10.1063/1.1610433 |
| [11] |
K. Ohshimo, K. Akimoto, M. Ogawa, et al., J. Phys. Chem. A 122 (2018) 2927-2932. DOI:10.1021/acs.jpca.8b00246 |
| [12] |
E. Fernández, M. Boronat, A. Corma, J. Phys. Chem. C 119 (2015) 19832-19846. DOI:10.1021/acs.jpcc.5b05023 |
| [13] |
S. Hirabayashi, M. Ichihashi, Y. Kawazoe, T. Kondow, J. Phys. Chem. A 116 (2012) 8799-8806. DOI:10.1021/jp304214m |
| [14] |
B.J. Winter, E.K. Parks, S.J. Riley, J. Chem. Phys. 94 (1991) 8618-8621. DOI:10.1063/1.460046 |
| [15] |
B. Yin, T. Wang, Y. Chen, et al., Int. J. Mass Spectrom. 451 (2020) 116312. DOI:10.1016/j.ijms.2020.116312 |
| [16] |
B. Yin, Z. Luo, Coord. Chem. Rev. 429 (2021) 213643. DOI:10.1016/j.ccr.2020.213643 |
| [17] |
J. Hagen, L.D. Socaciu, Roux J.Le, et al., J. Am. Chem. Soc. 126 (2004) 3442-3443. DOI:10.1021/ja038948r |
| [18] |
A.P. Woodham, G. Meijer, A. Fielicke, Angew. Chem. Int. Ed. 51 (2012) 4444-4447. DOI:10.1002/anie.201108958 |
| [19] |
T.M. Bernhardt, J. Hagen, S.M. Lang, et al., J. Phys. Chem. A 113 (2009) 2724-2733. DOI:10.1021/jp810055q |
| [20] |
S. Klacar, A. Hellman, I. Panas, H. Grönbeck, J. Phys. Chem. C 114 (2010) 12610-12617. DOI:10.1021/jp102715r |
| [21] |
B.E. Salisbury, W.T. Wallace, R.L. Whetten, Chem. Phys. 262 (2000) 131-141. DOI:10.1016/S0301-0104(00)00272-X |
| [22] |
T. Wang, J. Ma, B. Yin, X. Xing, J. Phys. Chem. A 122 (2018) 3346-3352. DOI:10.1021/acs.jpca.8b00629 |
| [23] |
J. Ma, X. Cao, X. Xing, X. Wang, J.H. Parks, Phys. Chem. Chem. Phys. 18 (2016) 743-748. DOI:10.1039/C5CP06116D |
| [24] |
W. Huang, H.J. Zhai, L.S. Wang, J. Am. Chem. Soc. 132 (2010) 4344-4351. DOI:10.1021/ja910401x |
| [25] |
R. Pal, L.M. Wang, Y. Pei, L.S. Wang, X.C. Zeng, J. Am. Chem. Soc. 134 (2012) 9438-9445. DOI:10.1021/ja302902p |
| [26] |
D. Stolcic, M. Fischer, G. Ganteför, et al., J. Am. Chem. Soc. 125 (2003) 2848-2849. DOI:10.1021/ja0293406 |
| [27] |
Z. Luo, G.U. Gamboa, J.C. Smith, et al., J. Am. Chem. Soc. 134 (2012) 18973-18978. DOI:10.1021/ja303268w |
| [28] |
A.C. Reber, S.N. Khanna, P.J. Roach, W.H. Woodward, A.W. Castleman, J. Am. Chem. Soc. 129 (2007) 16098-16101. DOI:10.1021/ja075998d |
| [29] |
L. Geng, B. Yin, H. Zhang, Z.D. Sun, Z. Luo, Nano. Res. 14 (2021) 4822-4827. DOI:10.1007/s12274-021-3435-7 |
| [30] |
H. Schwarz, Int. J. Mass Spectrom. 237 (2004) 75-105. DOI:10.1016/j.ijms.2004.06.006 |
| [31] |
R. Burgert, H. Schnöckel, A. Grubisic, et al., Science 319 (2008) 438. DOI:10.1126/science.1148643 |
| [32] |
H. Zhang, H. Wu, Y. Jia, et al., Rev. Sci. Instrum. 90 (2019) 073101. DOI:10.1063/1.5108994 |
| [33] |
J.J. Zhao, R.H. Xie, J. Comput. Theor. Nanosci. 1 (2004) 117-131. DOI:10.1166/jctn.2004.010 |
| [34] |
J. Zhao, X. Huang, R. Shi, et al., Chem. Model. 12 (2015) 249-292. |
| [35] |
B. Delley, J. Chem. Phys. 113 (2000) 7756-7764. DOI:10.1063/1.1316015 |
| [36] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [37] |
A.V. Krukau, O.A. Vydrov, A.F. Izmaylov, G.E. Scuseria, J. Chem. Phys. 125 (2006) 224106. DOI:10.1063/1.2404663 |
| [38] |
M. Frisch, G. Trucks, H. Schlegel, et al., Gaussian Inc., Wallingford CT, 2016.
|
| [39] |
M. Dolg, U. Wedig, H. Stoll, H. Preuss, J. Chem. Phys. 86 (1987) 866-872. DOI:10.1063/1.452288 |
| [40] |
R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72 (1980) 650-654. DOI:10.1063/1.438955 |
| [41] |
M.P. Mitoraj, A. Michalak, T. Ziegler, J. Chem. Theory Comput. 5 (2009) 962-975. DOI:10.1021/ct800503d |
| [42] |
G. te Velde, F.M. Bickelhaupt, E.J. Baerends, et al., J. Comput. Chem. 22 (2001) 931-967. DOI:10.1002/jcc.1056 |
| [43] |
E. J. Z. Baerends, T. Autschbach, J. Bashford, et al., http://www.scm.com.
|
| [44] |
B. Yin, Q. Du, L. Geng, et al., J. Phys. Chem. Lett. 11 (2020) 5807-5814. DOI:10.1021/acs.jpclett.0c01643 |
| [45] |
K.J. Taylor, C.L. Pettiette-Hall, O. Cheshnovsky, R.E. Smalley, J. Chem. Phys. 96 (1992) 3319-3329. DOI:10.1063/1.461927 |
| [46] |
B. Yin, Q. Du, L. Geng, et al., CCS Chem. 2 (2021) 219-229. |
| [47] |
M.M. Montemore, M.A. van Spronsen, R.J. Madix, C.M. Friend, Chem. Rev. 118 (2018) 2816-2862. DOI:10.1021/acs.chemrev.7b00217 |
| [48] |
H. Häkkinen, S. Abbet, A. Sanchez, U. Heiz, U. Landman, Angew. Chem. Int. Ed. 42 (2003) 1297-1300. DOI:10.1002/anie.200390334 |
| [49] |
M. Soustelle, Acid/Base Equilibria, in: Ionic and Electrochemical Equilibria, John Wiley & Sons, Inc., Hoboken, 2015, pp. 61-100.
|
| [50] |
J. Simons, P. Jorgensen, H. Taylor, O. Judy, J. Phys. Chem. 87 (1983) 2745-2753. DOI:10.1021/j100238a013 |