 2022, Vol. 33
2022, Vol. 33 


b School of Pharmacy and Bioengineering, Chongqing University of Technology, Chongqing 400054, China;
c Guangxi Key Laboratory of Chemistry and Engineering of Forest Products, Guangxi University for Nationalities, Nanning 530006, China;
d University of Chinese Academy of Sciences, Beijing 100049, China
Benzopyran-fused polycyclic ketals are prevalent in diverse natural products with significant biological properties such as potent L-calcium channel blocking activity, inhibitory activity against acetylcholine esterase, antioxidative property, and protective effect on mitochondria [1–5]. Their unique structural characteristics and potential biological activities have made them privileged pharmacophores for the development of industrially significant pharmaceuticals and agrochemicals. Moreover, their fascinating and complex scaffolds with potential usefulness, which were responsive for various serious diseases or pharmacological applications, have served as popular synthetic targets and sparked a wide range of notable synthetic efforts in recent years [6–22]. However, their access with high stereoselectivity under mild conditions remains to be desired. Herein, we report a bioinspired hemiacetalization/dehydration/[3 + 3]-type cycloaddition cascade to construct benzopyran-fused polycyclic ketals with generally good diastereoselectivities under mild conditions and the first bioinspired total syntheses of hyperaspidinols A (1) and B (2) via this strategy.
Hyperaspidinols A and B were isolated from Hypericum chinense L. (Hypericaceae) [23], which served as highly active traditional herb agents for the treatment of sepsis, acute laryngopharyngitis, conjunctivitis, hepatitis, and snake bite [24–28]. Structurally, hyperaspidinols A and B feature a novel furo[2,3-b]chromene framework (Fig. 1, highlighted in bold) and represent an intriguing and ubiquitous family of lignan derivatives with fascinating hybrid skeletons (Fig. 1) [23]. The remarkable potential biological activities and novel structural features of the two molecules make them appealing targets for the synthetic community [29–31]. Moreover, the successful syntheses of hyperaspidinols A and B will pave the way to efficiently access a series of biologically interesting natural products with hybrid structures such as melipatulinones A–C [32], patulignans (Fig. 1) [33] and divergent syntheses of their analogues towards drug development.

|
Download:
|
| Fig. 1. Representative natural products bearing furo[2,3-b]chromene or hybrid skeletons. | |
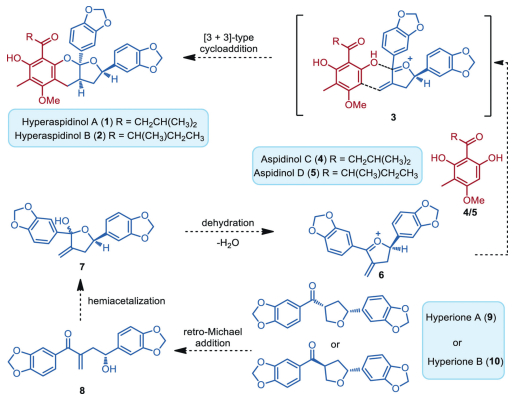
Inspired by these characteristic scaffolds in pharmaceutically active natural products, we speculated that hyperaspidinols A and B could evolve from substantially less complex natural products hyperione 9 or 10 by hybridization with the corresponding aspidinols C (4) and D (5), which were isolated together with hyperaspidinols A and B from the same herb [23]. Our continued interest in biomimetic construction of polycyclic ketal skeletons [34–38] guided us to speculate that hyperaspidinols A and B might arise from the methyleneoxonium intermediate 6 via a formal [3 + 3]-type cycloaddition cascade process (Scheme 1), similar to our previous strategies utilized in biomimetic total syntheses of myrtucommuacetalone [35], sanctis [37], and tomentosones [38]. 6 might generate from hyperione A (9) or B (10) via a retro-Michael addition/hemiacetalization/dehydration cascade sequence. In this regard, from the biogenetic perspective, if a one-pot acid-catalyzed cascade sequence merging the fascinating multistep transformations was established, it would render straightforward syntheses of hyperaspidinols A and B (1 and 2) with high efficiency.

|
Download:
|
| Scheme 1. Proposed biogenesis of hyperaspidinols A and B. | |
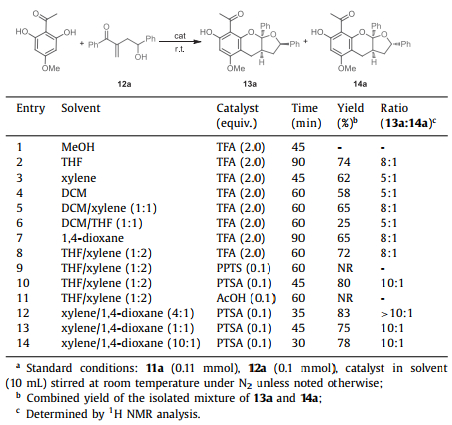
With the biomimetic scenario in mind, we first explored the construction of the benzopyran ketal skeleton. Optimization studies commenced with an evaluation of solvent, time, and catalyst by using the readily accessible phloroglucinol 11a [39,40] and hydroxyl vinyl ketone 12a [41] as model substrates (Table 1). When the two substrates were treated with TFA in methanol, only complex mixtures were observed (entry 1). Gratifyingly, switching the solvent to THF delivered the desired cascade reaction smoothly, furnishing the corresponding furo[2,3-b]chromene products 13a and 14a in 74% isolated yield with 8:1 diastereomeric ratio (entry 2).
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
{kind=link}
A systematic screening of solvents disclosed that nonpolar solvents such as xylene, dichloromethane, tetrahydrofuran, and their mixtures worsened the reaction in both yields and diastereoselectivities except for faster reaction rate (entries 2–6). The solvent screening showed that 1,4-dioxane and xylene/THF mixture solvents could provide almost identical yield and diastereomeric ratio as those of THF (entries 7 and 8). Attempting to further improve the yield and selectivity, we then screened several protonic acids as catalysts. Catalytic amount of pyridinium p-toluenesulfonate (PPTS) and stoichiometric AcOH were proven to be non-effective, whereas p-toluenesulfonic acid (PTSA) could trigger the desired transformation (entries 9–11). Notably, when we occasionally changed the solvent mixture to xylene/1,4-dioxane, both the yield and diastereoselectivity were obviously increased (entry 12). However, changing the ratio of the mixture solvents failed to further improve the reaction efficiency (entries 13 and 14). Finally, 0.1 equiv. of PTSA as catalyst in xylene/1,4-dioxane (4:1) were selected as the optimal reaction condition (83% yield, > 10:1 dr, Table 1, entry 12), thereby laying a solid foundation for the efficient syntheses of the benzopyran-fused polycyclic ketals.
With the optimized reaction conditions established, the scope of the biomimetic cascade reaction with respect to both reaction partners was evaluated. A series of hydroxyl vinyl ketones (12) could be converted to the corresponding furo[2,3-b]chromene products 13 and 14 in moderate to high yields with different diastereoselectivities as summarized in Scheme 2. Hydroxyl vinyl ketones bearing different aryl groups such as 4-methylphenyl (12b or 12e), 4-methoxylphenyl (12c), and 4-bromophenyl (12d) were suitable reaction candidates, delivering the corresponding products 13b-e in 55%–86% yields with good diastereoselectivities. Notably, hydroxyl vinyl ketones with an alkyl group attached on the α-carbon gave good yields but with poor diastereoselectivities (13f-g and 14f-g), probably due to steric effects. Furthermore, hydroxyl vinyl ketones bearing an extended chain could also undergo this transformation smoothly albeit with imperfect stereoselectivity, allowing the efficient construction of benzopyran-fused polycyclic ketals 13h-i and 14h-i.

|
Download:
|
| Scheme 2. Substrate scope with respect to hydroxyl vinyl ketones. Reaction conditions: 11a (0.22 mmol), 12 (0.2 mmol), PTSA (0.02 mmol) in xylene/1,4-dioxane (4:1, 10 mL) stirred at room temperature for 0.5–3 h; combined yields of the isolated mixture of 13 and 14; ratio (13:14) determined by 1H NMR unless noted otherwise. | |
{kind=link}
Encouraged by the aforementioned excellent results, further efforts towards verifying the substrate scope and versatility of the phenolic reaction partner 11 were also conducted. As shown in Scheme 3, the phenol substrates with different acyl substituents such as iso-valeryl (11j) and n-hexanoyl (11k) were well tolerated under the standard reaction conditions, which could give rise to the desired products in good yields and diastereomeric ratios, thus clarifying excellent tolerance of the developed cascade sequence. Similarly, the ester substituted phloroglucinol (11l) could also smoothly react with hydroxyl vinyl ketone 12a to afford the polycyclic ketals 13l and 14l in 70% overall yield with a ratio about 3:1. 3,5-Dimethoxyphenol 11m was also suitable reaction partner, allowing the synthesis of 13m and 14m in reasonable yield with acceptable diastereoselectivity. Moreover, diacylphloroglucinol 11n delivered the corresponding products 13n and 14n with comparable yield and diastereoselectivity to those of the monoacylphloroglucinol 11j. Much to our delight, the methyl substituted phloroglucinols (11o-q) could undergo this transformation and afford the corresponding furo[2,3-b]chromene products in yields more than 80% with good to excellent diastereoselectivities, generating a series of structurally diverse and closely similar analogues of melipatulinones A-C, which marked an advantage of this method and strongly indicated the promising utility of this cascade methodology to efficiently synthesize such lignin-phloroglucinol hybrid natural products and pharmacological compounds.

|
Download:
|
| Scheme 3. Substrate scope with respect to phenolic reaction partner. Reaction conditions: 11 (0.22 mmol), 12a (0.2 mmol), PTSA (0.02 mmol) in xylene/1,4-dioxane (4:1, 10 mL) stirred at room temperature for 0.5–3 h; combined yield of the isolated mixture of 13 and 14; ratio (13:14) determined by 1H NMR unless noted otherwise. aRatio of isolated products of 13 and 14. | |
{kind=link}
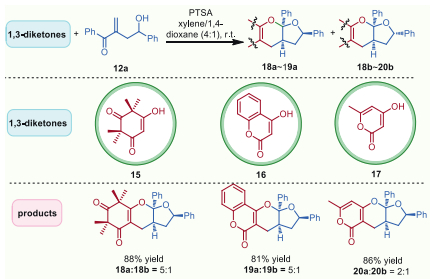
The synthetic possibility of this fascinating bioinspired hemiacetalization/dehydration/[3 + 3]-type cycloaddition cascade reaction was further reflected by intelligent orchestrations with straightforward structural combinations for the critical fragments of the pharmaceutically meaningful natural products. As delineated in Scheme 4, the potential bioactive fragment syncarpic acid 15, which shared an intriguing tetramethylcyclohexenedione skeleton responsive for the most characteristic unit of natural polycyclic polymethylated phloroglucinols mainly encountered in the Myrtaceae plants [42], was treated with hydroxyl vinyl ketone 12a, and the desired cascade reaction proceeded smoothly, delivering polycyclic ketals 18a and 18b in 88% yield with good diastereoselectivity. Moreover, 4-hydroxy-2H-chromen-2-one (16), answering for the biologically meaningful pharmaceutical core scaffold in numerous natural products [43,44] underwent the established reaction, also providing the desired products 19a and 19b in good combined yield as expected. Remarkably, the common naturally occurring 4-hydroxy-6-methyl-2-pyrone (17) [45] was also successfully converted to the targeted polycyclic ketals 20a and 20b in satisfactory yield. Collectively, the aforementioned representative structural combinations of the critical natural product fragments chemologically clarified its promising potential to efficiently access a focused library of natural products-modified analogues bearing diverse biologically functional units and might open up new vistas in structural optimizations of related natural products for broad scientific interest.

|
Download:
|
| Scheme 4. Substrate scope with respect to 1,3-diketones. Reaction conditions: 1,3-Diketone (0.22 mmol), 12a (0.2 mmol), PTSA (0.02 mmol) in xylene/1,4-dioxane (4:1, 10 mL) stirred at room temperature for 0.5–3 h; combined yield of diastereomers; ratio of the diastereomers. | |
{kind=link}
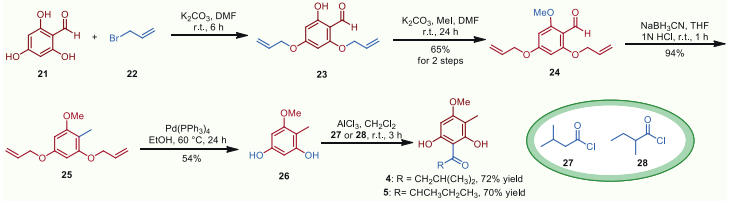
Building on the above initial success of the bioinspired methodology of hemiacetalization/dehydration/[3 + 3]-type cycloaddition cascade, the concise total syntheses of hyperaspidinols A and B were then performed to verify its potent synthetic utility. We initiated the preparation of the corresponding phloroglucinols 4 and 5 as depicted in Scheme 5. Selective protection of the free phenolic hydroxyl groups with allyl bromide followed by methylation of 2,4,6-trihydroxybenzaldehyde efficiently afforded aldehyde 24 in 65% yield over 2 steps. Further treatment of aldehyde 24 with NaBH3CN under acidic condition generated 25 in 94% yield efficiently. Deprotection of the allyl functionalities with Pd(PPh3)4 expectedly freed the two phenolic hydroxyl groups in moderate yield. Next, the critical phloroglucinol precursors 4 and 5 (asipidinols C and D) were delivered propitiously via Friedel-Crafts acylation of phenol 26 with acyl chlorides 27 and 28 in about 72% and 70% yield, respectively. Notably, asipidinols C (4) and D (5) were also biologically active natural products with various pharmaceutical activities [46,47] and they were completely synthesized for the first time in this study.

|
Download:
|
| 5. Syntheses of critical precursors asipidinols C and D (4, 5). | |
{kind=link}
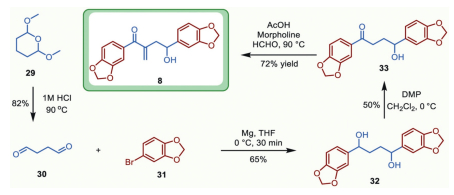
At this stage, efforts were further made to synthesize the other critical precursor 8. As shown in Scheme 6, 8 was readily prepared from commercially available acetal 29, which underwent hydrolysis under an acidic condition to afford butanedial 30 referring to previously established procedure [48]. Exposure of butanedial 30 to the Grignard reagent generated in situ from 4-bromo-1,2-(methyl enedioxy)benzene 31 and magnesium under nitrogen atmosphere smoothly delivered the symmetrical diol 32 in 65% yield. Selective mono-oxidation of benzyl diol 32 with Dess-Martin periodinane (DMP) furnished the targeted aryl ketone 33 in moderate yield. The morpholine catalyzed aldol condensation followed by dehydration afforded the critical precursor 8 in 72% yield.

|
Download:
|
| Scheme 6. Synthesis of critical precursor 8. | |
{kind=link}
Acquisition of the critical precursors asipidinols C and D (4, 5) and hydroxyl vinyl ketone 8 set the stage for the biomimetic total syntheses of hyperaspidinols A and B via the key biosynthetic hemiacetalization/dehydration/[3 + 3]-type cycloaddition cascade. Gratifyingly, aspidinol 4 reacted with hydroxyl vinyl ketone 8 smoothly to generate hyperaspidinol A in excellent yield with the diastereoslectivity more than 10:1, and the treatment of 8 with racemic aspidinol 5 gave the desired product in 92% yield as a 10:1 mixture of diastereoisomers favoring hyperaspidinol B (2) (Scheme 7). The NMR and HR-ESIMS spectra of the synthetic hyperaspidinol A are in full agreement with literature reports [23], and the synthetic hyperaspidinol B displayed NMR spectral properties in agreement with the reported ones except the different diastereomeric ratios (about 1:1) in regard to the acyl group on the phloroglucinol motif [23]. In this regard, the first total syntheses of hyperaspidinols A and B were accomplished, which strongly suggested that the aforementioned hemiacetalization/dehydration/[3 + 3]-type cycloaddition cascade sequence might probably proceed by mimicking their biosynthetic pathway.

|
Download:
|
| Scheme 7. Biomimetic total syntheses of hyperaspidinols A and B (1, 2). | |
{kind=link}
In summary, a biomimetic hemiacetalization/dehydration/[3 + 3]-type cycloaddition cascade methodology for the efficient establishment of furo[2,3-b]chromene was defined, and it was attested by the efficient total syntheses of hyperaspidinols A and B. The effectiveness of our strategy is borne out by the fact that a broad spectrum of novel furo[2,3-b]chromene derivatives could be constructed in moderate to good yields with different diastereoselectivities. Further applications of this strategy in the total syntheses of other natural products with similar skeletons are currently ongoing in our laboratory and will be reported in due course.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported financially by the National Natural Science Foundation of China (Nos. 81773602, 21901049 and 21801032), Natural Science Foundation of Guangdong Province (No. 2019A1515011694), Guangdong Special Support Program (No. 2017TQ04R599), Youth Innovation Promotion Association of CAS (No. 2020342), and Guangxi Natural Science Foundation (Nos. 2018GXNSFBA050015 and AD19245004).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.08.035.
| [1] |
Y.C. Lin, X.Y. Wu, S. Feng, et al., J. Org. Chem. 66 (2001) 6252-6256. DOI:10.1021/jo015522r |
| [2] |
X.Y. Wu, X.H. Liu, Y.C. Lin, et al., Eur. J. Org. Chem. 71 (2005) 4061-4064. DOI:10.1002/ejoc.200500326 |
| [3] |
J.D. Pettigrew, P.D. Wilson, Org. Lett. 8 (2006) 1427-1429. DOI:10.1021/ol060266w |
| [4] |
Z. Xu, Y. Li, Q. Xiang, et al., J. Med. Chem. 53 (2010) 4642-4653. DOI:10.1021/jm1001502 |
| [5] |
L. Feng, M.M. Maddox, M.Z. Alam, et al., J. Med. Chem. 57 (2014) 8398-8420. DOI:10.1021/jm500853v |
| [6] |
C. Airoldi, F. Cardona, E. Sironi, et al., Chem. Commun. 47 (2011) 10266-10268. DOI:10.1039/c1cc13046c |
| [7] |
Y. Luan, H. Sun, S.E. Schaus, Org. Lett. 13 (2011) 6480-6483. DOI:10.1021/ol202772k |
| [8] |
X.J. Lv, W.W. Zhao, Y.H. Chen, et al., Org. Chem. Front. 6 (2019) 1972-1976. DOI:10.1039/C9QO00366E |
| [9] |
Y.T. Lai, K. Nagaraju, R. Gurubrahamam, et al., Adv. Synth. Catal. 362 (2020) 3846-3850. DOI:10.1002/adsc.202000667 |
| [10] |
R. Rodriguez, R.M. Adlington, J.E. Moses, et al., Org. Lett. 6 (2004) 3617-3619. DOI:10.1021/ol048479d |
| [11] |
J.S. Yadav, B.V.S. Reddy, P.N. Reddy, Chem. Lett. 33 (2004) 1436-1437. DOI:10.1246/cl.2004.1436 |
| [12] |
W. Luo, Q.S. Yu, M. Zhan, et al., J. Med. Chem. 48 (2005) 986-994. DOI:10.1021/jm049309+ |
| [13] |
B. Biswas, D. Sarkar, R.V. Venkateswaran, Tetrahedron 64 (2008) 3212-3216. DOI:10.1016/j.tet.2008.01.097 |
| [14] |
D.L. Paterson, D. Barker, Beilstein J. Org. Chem. 11 (2015) 265-270. DOI:10.3762/bjoc.11.29 |
| [15] |
J. Yang, G. Qiu, J. Jiang, et al., Adv. Synth. Catal. 359 (2017) 2184-2190. DOI:10.1002/adsc.201601335 |
| [16] |
J. Guo, H. Miao, Y. Zhao, et al., Chem. Commun. 55 (2019) 5207-5210. DOI:10.1039/C9CC02170A |
| [17] |
L. Zhou, W.G. Yan, X.L. Sun, et al., Angew. Chem. Int. Ed. 59 (2020) 18964-18969. DOI:10.1002/anie.202007068 |
| [18] |
J. Wandji, S.S. Awanchiri, Z.T. Fomum, et al., Phytochemistry 38 (1995) 1309-1313. DOI:10.1016/0031-9422(94)00671-F |
| [19] |
K. Krohn, M. Riaz, Tetrahedron Lett. 45 (2004) 293-294. DOI:10.1016/j.tetlet.2003.10.189 |
| [20] |
J.D. Pettigrew, P.D. Wilson, J. Org. Chem. 71 (2006) 1620-1625. DOI:10.1021/jo052371+ |
| [21] |
B. Panda, T.K. Sarkar, J. Org. Chem. 78 (2013) 2413-2421. DOI:10.1021/jo302545n |
| [22] |
B. Roy, N. Rout, P. Kuila, et al., J Heterocyclic Chem. 58 (2021) 8-27. DOI:10.1002/jhet.4152 |
| [23] |
W. Wang, Y.H. Zeng, K. Osman, et al., J. Nat. Prod. 73 (2010) 1815-1820. DOI:10.1021/np1004483 |
| [24] |
S. Gibbons, B. Ohlendorf, I. Johnsen, Fitoterapia 73 (2002) 300-304. DOI:10.1016/S0367-326X(02)00082-5 |
| [25] |
X. Jia, Y. Wu, C. Lei, et al., Chin. Chem. Lett. 31 (2020) 1263-1266. DOI:10.1016/j.cclet.2019.10.014 |
| [26] |
Y. Ye, N. Jiang, X. Yang, et al., Chin. Chem. Lett. 31 (2020) 2433-2436. DOI:10.1016/j.cclet.2020.04.028 |
| [27] |
J. Ma, G. Xia, Y. Zang, et al., Chin. Chem. Lett. 32 (2021) 1173-1176. DOI:10.1016/j.cclet.2020.07.037 |
| [28] |
B. Zhen, X. Suo, J. Dang, et al., Chin. Chem. Lett. 32 (2021) 2338-2341. DOI:10.1016/j.cclet.2020.10.027 |
| [29] |
N. Duhamel, C.E. Rye, D. Barker, Asian J. Org. Chem. 2 (2013) 491-493. DOI:10.1002/ajoc.201300086 |
| [30] |
M.C. Sapu, J. Deska, Org. Biomol. Chem. 11 (2013) 1376-1382. DOI:10.1039/c2ob27073k |
| [31] |
W.D. Liang, X.P. Cai, M.J. Dai, Chem. Sci. 12 (2012) 1311-1316. |
| [32] |
V.T. Vu, X.L. Chen, L.Y. Kong, et al., Org. Lett. 22 (2020) 1380-1384. DOI:10.1021/acs.orglett.9b04680 |
| [33] |
V.T. Vu, M.T. Nguyen, W. Wang, et al., Org. Biomol. Chem. 18 (2020) 6607-6611. DOI:10.1039/D0OB01412E |
| [34] |
H. Tan, H. Liu, X. Chen, et al., Org. Lett. 17 (2015) 4050-4053. DOI:10.1021/acs.orglett.5b01970 |
| [35] |
H. Liu, L. Huo, B. Yang, et al., J. Org. Lett. 19 (2017) 4786-4789. DOI:10.1021/acs.orglett.7b02159 |
| [36] |
H. Liu, W. Yu, X. Guo, et al., Org. Lett. 20 (2018) 546-549. DOI:10.1021/acs.orglett.7b03630 |
| [37] |
L. Huo, C. Dong, M. Wang, et al., Org. Lett. 22 (2020) 934-938. DOI:10.1021/acs.orglett.9b04486 |
| [38] |
X. Zhang, C. Dong, G. Wu, et al., Org. Lett. 22 (2020) 8007-8011. DOI:10.1021/acs.orglett.0c02943 |
| [39] |
D.H. Dethe, A.K. Nirpal, Org. Biomol. Chem. 17 (2019) 7507-7516. DOI:10.1039/C9OB01426H |
| [40] |
A.J. Grenning, J.H. Boyce, J.A. Porco Jr, J. Am. Chem. Soc. 136 (2014) 11799-11804. DOI:10.1021/ja5060302 |
| [41] |
E. Jung, N. Dittrich, L.I. Pilkington, et al., Tetrahedron 71 (2015) 9439-9456. DOI:10.1016/j.tet.2015.10.050 |
| [42] |
O. Celaj, A.G. Durán, P. Cennamo, et al., Phytochem. Rev. 20 (2021) 259-299. DOI:10.1007/s11101-020-09697-2 |
| [43] |
D.S. Reddy, M. Kongot, A. Kumar, Tuberculosis 127 (2021) 102050. DOI:10.1016/j.tube.2020.102050 |
| [44] |
Z. Xu, Q.T. Chen, Y. Zhang, et al., Fitoterapia 150 (2021) 104863. DOI:10.1016/j.fitote.2021.104863 |
| [45] |
G.P. McGlacken, I.J.S. Fairlam, Nat. Prod. Rep. 22 (2005) 369-385. DOI:10.1039/b416651p |
| [46] |
H.X. Liu, Y.C. Chen, Y. Liu, et al., Fitoterapia 114 (2016) 40-44. DOI:10.1016/j.fitote.2016.08.010 |
| [47] |
V. Brezáni1, V. Leláková, S. Hassan, et al., Viruses 10 (2018) 360. DOI:10.3390/v10070360 |
| [48] |
S. Prévost, K. Thai, N. Schützenmeister, et al., Org. Lett. 17 (2015) 504-507. DOI:10.1021/ol503520f |