 2022, Vol. 33
2022, Vol. 33 


In terms of the high efficiency and good atom-/step-economy, transition metal (TM)-catalyzed C-H functionalization strategy has been demonstrated as a practical protocol in synthetic chemistry [1-3]. Up to date, diversified directing groups (DGs) and coupling partners (CPs) have been developed to achieve challenging reaction modes in this field [4-11]. On the other hand, the cascade reaction involving multiple new bonds formation via a simple one-pot operation has drawn continuous interest due to its superiority in assembling complex molecules, consequently, the combination of TM-catalyzed C-H functionalization and the cascade strategy has been proven to be a powerful approach for the rapid synthesis of polycyclic ring-fused motifs [12-18]. Azomethine imines, equipped with a dipolar fragment, had been developed as versatile reactants to participate in diverse C-H functionalization/dipolar cycloaddition cascades, which provided reliable synthetic methods to afford various ring-fused skeletons (Scheme 1a) [19-24]. Despite these remarkable advances, there is still room for improvement, e.g., developing novel reaction manifold and realizing asymmetric synthesis of intriguing complex structures.

|
Download:
|
| Scheme 1. Diversified assembly of ring-fused motifs via TM-catalyzed C-H functionalization of azomethine imines. | |
More recently, by appropriately modifying the atropchiral cyclopentadiene (Cp) ligands, a variety of CpX-coordinated rhodium(Ⅲ) (CpXRh(Ⅲ)) complexes have been developed as competent catalysts to fulfill asymmetric C-H functionalization reactions [25-28]. Compared with precedented CpXRh(Ⅲ)-catalyzed C-H couplings [29-36], it is changeling, but extremely attractive to explore the versatility of this strategy for constructing chiral polycyclic ring-fused frameworks via cascade transformations. To address this issue: 1) a TM-catalyzed cascade process is typically involved in forming the chiral center; 2) proper coordinated environments should be engaged in balancing the reactivity and stereoselectivity. Taking advantage of chiral CpXRh(Ⅲ)-catalyzed asymmetric C-H functionalization in fulfilling intriguing enantioenriched skeletons, herein, we would like to disclose the enantioselective C-H couplings of azomethine imines with propargyl carbonates [37-41]for the construction of chiral tetracyclic indenopyrazolopyrazolone derivatives (Scheme 1b). This transformation was rationally designed on the basis of Rh(Ⅲ)-catalyzed stepwise cycloaddition process, which was probed by detailed DFT calculations. Further rationalization of the enantioselectivity and the role of dual directing groups (DDGs, azomethine imine and the –OBoc moiety) in tuning the selectivity were also clarified.
At the outset of our investigation, the racemic C-H coupling of azomethine imine 1a with propargyl carbonate 2a was successfully realized in the presence of diverse TM catalysts, giving access to the specific assembly of tetracyclic indenopyrazolopyrazolone framework 3aa (Table S1 in Supporting information) [42]. Both ruthenium(Ⅱ)- and Rh(Ⅲ)-catalytic systems were demonstrated to be ideal for this transformation, while iridium(Ⅲ) catalyst gave relatively inferior efficiency. Encouraged by these results and in consideration of the Cp*Rh(Ⅲ)-enabled diverse novel reaction manifolds, we were next intrigued to gain more insight into the reaction mechanism, in particular, to clarify the potential synergy directing mode of azomethine imine and the -OBoc moiety with the catalyst metal in tuning both the reactivity and regioselectivity, thus providing an opportunity to realize the enantioselective synthesis of the corresponding ring-fused motifs.
In continuation of our work on exploring the synergistic coordination mode of DDGs in Rh(Ⅲ)-catalyzed C-H functionalization [43-45], therefore, a set of density functional theory (DFT) studies were first carried out by selecting the rhodacycle INT-0 as the starting point with TFE being the solvent. Initially, the coordination of propargyl carbonate with rhodium metal (INT-1) followed by regioselective insertion of the alkyne into C-Rh bond via the transition state TS-1 (∆G≠ = 21.4 kcal/mol) led to the formation of INT-3, which facilely converted into a more stable intermediate INT-4 bearing distinct coordination affinity between -OBoc and the rhodium metal center (Fig. 1). Thereafter, the β-O elimination from INT-4 proceeded via TS-3 with an energy barrier of 7.6 kcal/mol, giving the allene intermediate INT-5 with an exothermic process (path Ⅰ). Different possible pathways for the following [3 + 2] cycloaddition were then calculated. In path Ⅰa, a metal-free annulation from INT-6c proceeded through TS-4c with an energy barrier of 24.6 kcal/mol (from INT-5 to TS-4c), affording the final tetracyclic indenopyrazolopyrazolone PC with an overall exothermicity by 33.8 kcal/mol. As a comparison, the Rh(Ⅲ)-catalyzed [3 + 2] cycloaddition via TS-4 (∆G≠ = -1.8 kcal/mol) and TS-5 (∆G≠ = -9.8 kcal/mol) led to the formation of PC with a relatively lower energy barrier of 13.7 kcal/mol (path Ⅰb, from INT-5 to TS-4), implying the rhodium metal might be involved in the annulation process. On the other hand, removal of the -OBoc moiety from INT-3 resulted in the coordination of the allene double bond with rhodium metal (INT-6b), followed by the annulation through TS-4b with an energy barrier of 24.5 kcal/mol (path Ⅰc). Obviously, path Ⅰb involving the Rh(Ⅲ)-catalyzed stepwise C-C/C-N bond formations was more reasonable based on the reaction energy profile, and -OBoc played a key role in tuning the regioselectivity and reactivity by the coordination interaction. An alternative Rh(Ⅲ)-catalyzed stepwise C-N/C-C formation process involving 8-membered rhodacycle species was ruled out due to the high energy barriers (Fig. S1 in Supporting information).

|
Download:
|
| Fig. 1. Computed Gibbs free energy changes of the reaction pathways. | |
Alternatively, the intramolecular nucleophilic addition of the vinyl C-Rh bond in INT-4 onto the iminium via TS-3a led to the formation of INT-5a with an energy barrier of 15.8 kcal/mol (path Ⅱ), further C-O bond cleavage via TS-4a (∆G≠ = -15.1 kcal/mol) offered the intermediate INT-6. Overall, the comparison of the energy barriers in different reaction pathways revealed that the cascade β-O elimination/Rh(Ⅲ)-catalyzed stepwise annulation via path Ⅰb was more favorable rather than that of path Ⅱ (13.7 vs. 15.8 kcal/mol), nevertheless, the relatively small energy difference of 2.1 kcal/mol dropped a hint that both two paths might be involved in the developed transformation as a competitive process. In addition, the Ru(Ⅱ)-catalyzed reaction paths were also calculated and featured a similar energy barrier of 13.0 kcal/mol for the cascade β-O elimination/intramolecular annulation, while relatively higher energy barrier (∆G≠ = 24.1 kcal/mol, from INT-4Ru to TS-3aRu) was involved for the nucleophilic addition process in comparison with Rh(Ⅲ) catalysis (Fig. S2 in Supporting information). The results demonstrated that different reaction pathways might be involved varying with the catalyst metal, in which the cascade C-H activation/β-O elimination/annulation process was more reasonable for Ru(Ⅱ)-catalyzed transformation.
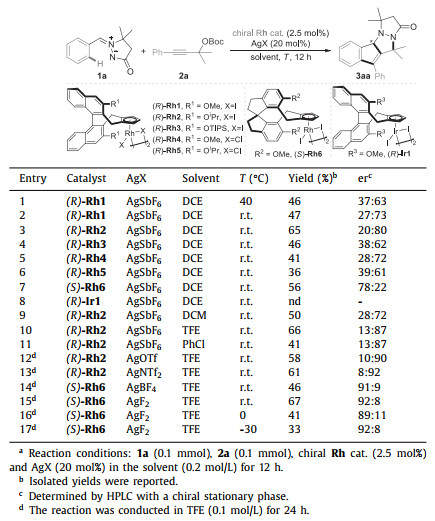
On the basis of the DFT calculation results and taking advantage of the revealed Cp*Rh(Ⅲ)-catalyzed [3 + 2] stepwise cycloaddition process, we envisioned that the rational design of a chiral CpXRh(Ⅲ) catalyst may enable the enantioselective pattern of the developed C-H cascades to furnish enantioenriched tetracyclic indenopyrazolopyrazolones. The asymmetric reaction of 1a with 2a was tested using a commercially available chiral (R)-Rh1 catalyst. As predicted, the desired enantioenriched product 3aa was obtained smoothly albeit with less satisfactory enantioselectivity (Table 1, entry 1). Further examination of other chiral Rh(Ⅲ) catalysts revealed that the -OiPr substituted (R)-Rh2 and the spiro Cp coordinated (S)-Rh6 resulted in relatively high er values, while chiral Ir(Ⅲ) catalyst (R)-Ir1 showed no reactivity under the similar conditions (entries 2-8). A screening of diverse reaction solvent indicated that TFE was optimal. Variation of diverse silver salts, additives and reaction concentrations demonstrated that the (R)-Rh2/AgNTf2 and (S)-Rh6/AgF2 system worked equally to furnish both enantiomers of 3aa with 92:8 er. Additionally, no more enhancement of the enantioselectivity was achieved by further reducing the reaction temperature down to -30 ℃ or employing the chiral Brønsted acid as a co-catalyst (Table S4 in Supporting information).
|
|
Table 1 Optimization of asymmetric reaction.a |
{kind=link}
{kind=link}
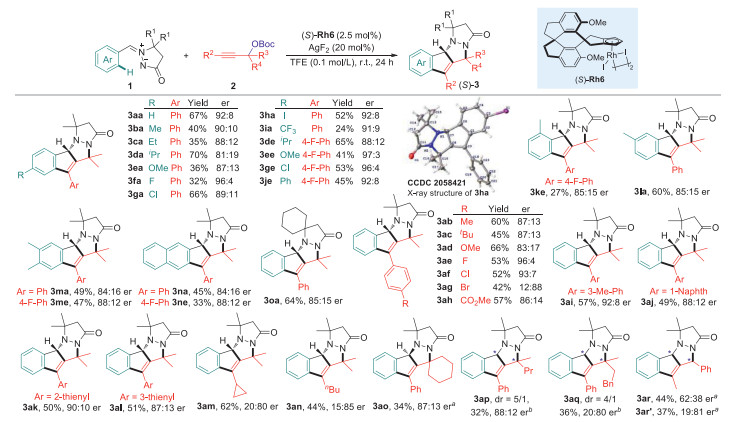
The scope of the chiral Rh(Ⅲ)-catalyzed asymmetric C-H coupling/[3 + 2] stepwise cycloaddition reaction was then examined under the optimal conditions in the presence of (S)-Rh6, giving a direct access to a variety of enantioenriched tetracyclic indenopyrazolopyrazolone products. As shown in Scheme 2, the chiral Rh(Ⅲ)-catalyzed C-H cascades were compatible to various azomethine imines regardless of the electronic properties and positions of the substitution on the phenyl ring, affording the corresponding indenopyrazolopyrazolones in good to excellent enantioselectivities (range from 81:19 to 97:3 er). The absolute configuration of the chiral carbon center was determined to be (S) by the X-ray crystallographic analysis of compound 3ha. Further exploration of the scope with regard to propargyl carbonates showed comparably good compatibility with aryl-, heteroaryl- or alkyl-substituted substrates, which reacted smoothly with 1a to provide the desired C-H alkenylation/[3 + 2] stepwise cycloaddition products (3ab-ao) in an enantioselective manner. Nevertheless, terminal alkyne and ester or pyridyl tethered propargyl carbonates were not compatible, thus illustrating the substrate limitation of this transformation. Of note, the use of asymmetrical propargyl carbonates (rac)-2p and (rac)-2q resulted in the formation of the tetracyclic indenopyrazolopyrazolone bearing two chiral centers, and the dr value was determined to be 5/1 and 4/1. When tert-butyl(1-phenylbut-2-yn-1-yl)carbonate was used, two diastereomers were also successfully obtained albeit with relatively moderate enantioselectivity (3ar and 3ar').

|
Download:
|
| Scheme 2. Scope for the enantioselective synthesis of tetracyclic indenopyrazolopyrazolone derivatives. Reaction conditions: 1 (0.1 mmol), 2 (0.1 mmol), (S)-Rh6 (2.5 mol%) and AgF2 (20 mol%) in TFE (0.1 mol/L) at room temperature for 24 h, isolated yields were reported, the er values were determined by HPLC with a chiral stationary phase. aThe reaction was conducted in the presence of (R)-Rh2 (2.5 mol%) and AgNTf2 (20 mol%). bThe dr value was determined by 1H NMR analysis of crude products, the yield and the er value of the major isomer was reported. | |
{kind=link}
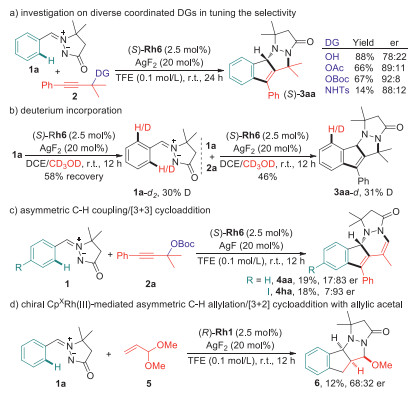
In combination with the computational mechanistic studies and the chiral CpXRh(Ⅲ)-enabled enantioselective pattern of the developed protocol, we were next intrigued to carry out a set of experimental mechanistic studies for deeper understanding of the DDGs-assisted strategy. Initially, diverse propargyl carbonate analogs were subjected to the (S)-Rh6 catalyzed conditions. The results showed that -OH, -OAc and –NHTs were all compatible coordinated DGs to enable the observed C-H alkenylation/[3 + 2] cycloaddition cascade, while er values varied with different steric hindrances. These results implied that the -OBoc moiety was essential in realizing high enantioselectivity of this reaction (Scheme 3a). Subsequently, deuterium-labeling experiment was conducted to figure out the reversibility of the C-H bond cleavage process. With CD3OD being the deuterium source, obvious deuterium incorporation at the ortho-position of DG in both recovered 1a and the tetracyclic indenopyrazolopyrazolone 3aa was detected, suggesting a reversible and fast C-H metalation process (Scheme 3b) [46]. In addition, the alternative asymmetric C-H alkenylation/[3 + 3] cycloaddition process occurred under the catalysis of (R)-Rh6/AgF, furnishing the corresponding tetracyclic indenopyrazolopyridazinone derivative 4 in an enantioselective manner, which provided a circumstantial evidence to support the formation of the allene intermediate followed by the allylic C-H bond cleavage mediated by Rh(Ⅲ) catalyst (Scheme 3c). In a comparison with precedented Ru(Ⅱ)-catalyzed C-H coupling of azomethine imine with allylic acetal [21], we tested the chiral CpXRh(Ⅲ) catalytic system for this transformation and delivered the desired product 6 with 68:32 er (Scheme 3d). This result demonstrated that the Rh(Ⅲ) catalyst should be involved in the [3 + 2] dipolar cycloaddition process rather than the previously reported metal-free annulation, which was in good line with our DFT calculations.

|
Download:
|
| Scheme 3. Experimental mechanistic studies. | |
{kind=link}
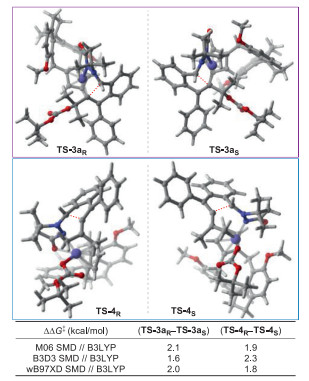
Further rationalization of the enantioselectivity in (S)-Rh6 catalyzed (S)-selective C-H coupling/[3 + 2] stepwise cycloaddition was carried out by DFT studies. On the basis of the computed Gibbs free energy change profiles shown in Fig. 1, the Rh(Ⅲ)-catalyzed annulation via TS-4 constituted the stereo-determining step of path Ⅰb, while the neucleophilic addition through TS-3a constituted the stereo-determining step of path Ⅱ. Accordingly, the transition states TS-4R/TS-4S and TS-3aR/TS-3aS with respect to both (R)- and (S)-enantiomers via different reaction pathways were investigated and the differences of free energies were listed in Fig. 2. The calculations using different levels of theoretical studies showed consistent results that the free energy of TS-4R/TS-3aR was higher than that of the corresponding (S)-enantiomer, with the energy difference ranged from 1.6 kcal/mol to 2.3 kcal/mol, which accounted for our experimental observation that the enantioenriched (S)-3 were formed enantioselectively. The energy barriers for the assembly of (S)-3aa via both (S)-Rh6-catalyzed paths were also calculated (Fig. S3 in Supporting information) and resulted in a value of 15.2 kcal/mol for path Ⅰb (from INT-5S to TS-4S) and 14.1 kcal/mol for path Ⅱ (from INT-4S to TS-3aS). The small energy difference of 1.1 kcal/mol illustrated a competitive process in this transformation, which might be the origin of relatively moderate er values for some substrates due to the difficulty in tuning both reaction paths enantioselectively.

|
Download:
|
| Fig. 2. Optimized geometries and energy differences of the transition states in the stereo-determining step for chiral (S)-Rh6 catalyzed tandem C-H alkenylation/[3 + 2] stepwise cycloaddition. | |
{kind=link}
On the basis of combined DFT calculations and experimental mechanistic studies, the catalytic cycle is tentatively proposed for the developed chiral CpXRh(Ⅲ)-catalyzed enantioselective transformation (Scheme 4). Initially, the cationic Rh(Ⅲ) species is formed as an active catalyst, followed by the azomethine imine-assisted reversible C-H bond activation to afford the six-membered rhodacycle A. Subsequent regioselective migratory insertion of the alkyne moiety delivers intermediate B, which features distinct coordination affinity between -OBoc and the rhodium metal center. Thereafter, two competitive pathways involving β-O elimination/Rh-catalyzed stepwise intramolecular annulation (path Ⅰ) or nucleophilic addition/C-N annulation (path Ⅱ) processes are realized to furnish the tetracyclic indenopyrazolopyridazinone (S)-3aa in a redox-neutral manner. The observed (S)-selectivity was rationalized by the model presented in Fig. 2 for both paths, in which the synergistic DDGs-assisted coordination mode plays crucial role in tuning the enantioselectivity.

|
Download:
|
| Scheme 4. Proposed catalytic cycle. | |
{kind=link}
In summary, we have developed the Rh(Ⅲ)-catalyzed and DDGs-assisted enantioselective C-H coupling/intramolecular cycloaddition cascade for the asymmetric synthesis of ring-fused tetracyclic indenopyrazolopyrazolone structures. Through integrated computational and experimental mechanistic studies, the detailed reaction pathway involving a catalyst metal-catalyzed stepwise annulation, the coordination mode and role of DDGs, as well as the origin of regio- and enantioselectivity have been elucidated. Further development of innovative synergistic DDGs-enabled C-H functionalization strategy for the construction of intriguing skeletons and biologically important organic building blocks are ongoing in our laboratory.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgmentsWe thank the National Natural Science Foundation of China (NSFC, Nos. 21877020, 22007020), Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2017A030306031) and Natural Science Foundation of Guangdong Province (No. 2019A1515010935) for financial support on this study.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.08.004.
| [1] |
H. Ito, K. Ozaki, K. Itami, Angew. Chem. Int. Ed. 56 (2017) 11144-11164. DOI:10.1002/anie.201701058 |
| [2] |
L. Li, Z. Chen, X. Zhang, Y. Jia, Chem. Rev. 118 (2018) 3752-3832. DOI:10.1021/acs.chemrev.7b00653 |
| [3] |
J. Bosson, J. Gouin, J. Lacour, Chem. Soc. Rev. 43 (2014) 2824-2840. DOI:10.1039/c3cs60461f |
| [4] |
S.M. Khake, N. Chatani, Chem. 6 (2020) 1056-1081. |
| [5] |
Q. Shao, K. Wu, Z. Zhuang, S. Qian, J.Q. Yu, Acc. Chem. Res. 53 (2020) 833-851. DOI:10.1021/acs.accounts.9b00621 |
| [6] |
W. Zhu, T.B. Gunnoe, Acc. Chem. Res. 53 (2020) 920-936. DOI:10.1021/acs.accounts.0c00036 |
| [7] |
A. Trowbridge, S.M. Walton, M.J. Gaunt, Chem. Rev. 120 (2020) 2613-2692. DOI:10.1021/acs.chemrev.9b00462 |
| [8] |
T. Jiang, H. Zhang, Y. Ding, et al., Chem. Soc. Rev. 49 (2020) 1487-1516. DOI:10.1039/c9cs00539k |
| [9] |
Q. Zhao, G. Meng, S.P. Nolan, M. Szostak, Chem. Rev. 120 (2020) 1981-2048. DOI:10.1021/acs.chemrev.9b00634 |
| [10] |
S. Rej, Y. Ano, N. Chatani, Chem. Rev. 120 (2020) 1788-1887. DOI:10.1021/acs.chemrev.9b00495 |
| [11] |
K.J. Jiao, Y.K. Xing, Q.L. Yang, H. Qiu, T.S. Mei, Acc. Chem. Res. 53 (2020) 300-310. DOI:10.1021/acs.accounts.9b00603 |
| [12] |
M.I.D. Mardjan, A. Mayooufi, J.L. Parrain, J. Thibonnet, L. Commeiras, Org. Process Res. Dev. 24 (2020) 606-614. DOI:10.1021/acs.oprd.9b00438 |
| [13] |
C. Zhu, H. Yue, L. Chu, M. Rueping, Chem. Sci. 11 (2020) 4051-4064. DOI:10.1039/d0sc00712a |
| [14] |
X.Y. Liu, Y. Qin, Acc. Chem. Res. 52 (2019) 1877-1891. DOI:10.1021/acs.accounts.9b00246 |
| [15] |
C.T. Walsh, B.S. Moore, Angew. Chem. Int. Ed. 58 (2019) 6846-6879. DOI:10.1002/anie.201807844 |
| [16] |
P. Gandeepan, T. Muller, D. Zell, et al., Chem. Rev. 119 (2019) 2192-2452. DOI:10.1021/acs.chemrev.8b00507 |
| [17] |
S.V. Kumar, S. Banerjee, T. Punniyamurthy, Org. Chem. Front. 7 (2020) 1527-1569. |
| [18] |
W. Yao, Y. Zhang, H. Zhu, C. Ge, D. Wang, Chin. Chem. Lett. 31 (2020) 701-705. |
| [19] |
W. Zhen, F. Wang, M. Zhao, Z. Du, X. Li, Angew. Chem. Int. Ed. 51 (2012) 11819-11823. DOI:10.1002/anie.201207204 |
| [20] |
D. Bai, T. Xu, C. Ma, et al., ACS Catal. 8 (2018) 4194-4200. DOI:10.1021/acscatal.8b00746 |
| [21] |
H. Lee, D. Kang, S.H. Han, et al., Angew. Chem. Int. Ed. 58 (2019) 9470-9474. DOI:10.1002/anie.201903983 |
| [22] |
Y. Cheng, X. Han, J. Li, Y. Zhou, H. Liu, Org. Chem. Front. 7 (2020) 3186-3192. DOI:10.1039/d0qo00786b |
| [23] |
Z. Shu, J. Zhou, J. Li, et al., J. Org. Chem. 85 (2020) 12097-12107. DOI:10.1021/acs.joc.0c01228 |
| [24] |
M. Wu, R. Wang, F. Chen, et al., Org. Lett. 22 (2020) 7152-7157. DOI:10.1021/acs.orglett.0c02506 |
| [25] |
B. Ye, N. Cramer, Acc. Chem. Res. 48 (2015) 1308-1318. DOI:10.1021/acs.accounts.5b00092 |
| [26] |
C.G. Newton, S.G. Wang, C.C. Oliveira, N. Cramer, Chem. Rev. 117 (2017) 8908-8976. DOI:10.1021/acs.chemrev.6b00692 |
| [27] |
J. Mas-Roselló, A.G. Herraiz, B. Audic, A. Laverny, N. Cramer, Angew. Chem. Int. Ed. 60 (2021) 13198-13224. DOI:10.1002/anie.202008166 |
| [28] |
T. Yoshino, S. Satake, S. Matsunaga, Chem. Eur. J. 26 (2020) 7346-7357. DOI:10.1002/chem.201905417 |
| [29] |
L. Wu, H. Xu, H. Gao, et al., ACS Catal. 11 (2021) 2279-2287. DOI:10.1021/acscatal.0c04777 |
| [30] |
L. Sun, H. Chen, B. Liu, et al., Angew. Chem. Int. Ed. 60 (2021) 8391-8395. DOI:10.1002/anie.202012932 |
| [31] |
Q. Wang, W.W. Zhang, C. Zheng, Q. Gu, S.L. You, J. Am. Chem. Soc. 143 (2021) 114-120. |
| [32] |
Q. Wang, W.W. Zhang, H. Song, et al., J. Am. Chem. Soc. 142 (2020) 15678-15685. DOI:10.1021/jacs.0c08205 |
| [33] |
F. Wang, Z. Qi, Y. Zhao, et al., Angew. Chem. Int. Ed. 59 (2020) 13288-13294. DOI:10.1002/anie.202002208 |
| [34] |
L. Kong, X. Han, S. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 7188-7192. DOI:10.1002/anie.202000174 |
| [35] |
G. Li, X. Yan, J. Jiang, et al., Angew. Chem. Int. Ed. 59 (2020) 22436-22440. DOI:10.1002/anie.202010489 |
| [36] |
C. Duchemin, N. Cramer, Angew. Chem. Int. Ed. 59 (2020) 14129-14133. DOI:10.1002/anie.202006149 |
| [37] |
Y. Yang, K. Zhang, J. Yang, et al., Chem. Commun. 56 (2020) 11315-11318. DOI:10.1039/d0cc04041j |
| [38] |
X. Song, Q. Zhou, J. Zhao, et al., Org. Lett. 22 (2020) 9506-9512. DOI:10.1021/acs.orglett.0c03515 |
| [39] |
Q. Lu, S. Mondal, S. Cembellín, S. Greßies, F. Glorius, Chem. Sci. 10 (2019) 6560-6564. DOI:10.1039/c9sc01703h |
| [40] |
G. Zheng, J. Sun, Y. Xu, S. Zhai, X. Li, Angew. Chem. Int. Ed. 58 (2019) 5090-5094. DOI:10.1002/anie.201900166 |
| [41] |
S. Wu, X. Wu, C. Fu, S. Ma, Org. Lett. 20 (2018) 2831-2834. DOI:10.1021/acs.orglett.8b00780 |
| [42] |
L. Zhang, J. Zhao, Y. Jiang, X. Zhang, X. Fan, Org. Chem. Front. 8 (2021) 3734-3739. DOI:10.1039/d1qo00025j |
| [43] |
G. Zheng, Z. Zhou, G. Zhu, et al., Angew. Chem. Int. Ed. 59 (2020) 2890-2896. DOI:10.1002/anie.201913794 |
| [44] |
W. Yi, W. Chen, F.X. Liu, et al., ACS Catal. 8 (2018) 9508-9519. DOI:10.1021/acscatal.8b02402 |
| [45] |
W. Gong, Z. Zhou, J. Shi, et al., Org. Lett. 20 (2018) 182-185. DOI:10.1021/acs.orglett.7b03546 |
| [46] |
M. Simonetti, G.J.P. Perry, X.C. Cambeiro, et al., J. Am. Chem. Soc. 138 (2016) 3596-3606. DOI:10.1021/jacs.6b01615 |