 2022, Vol. 33
2022, Vol. 33 


Since the first example of all-inorganic perovskites (AIP) nanomaterials were reported in 2015 [1], researchers have prepared a series of AIP with excellent photoelectric performances [2-5]. Although the photoluminescence quantum yield (PLQY) of AIP has reached near-unity [6], the low charge-transport property caused by non-conjugated organic ligands still makes a certain gap between AIP and traditional semiconductor quantum dots (QDs) [7] and organic luminescent materials [8], which seriously hindered the development level of AIP in many application field [9]. The ligand exchange reactions in polar solvents were used to improve the charge-transport property of traditional semiconductor QDs [10], which was useless for AIP with low stability in the polar solvents [11]. Therefore, researching effective methods for improving the charge-transport property of the AIP is imperative to promote the application of AIP.
In recent years, researchers have actively explored methods to improve the charge-transport property of AIP and have made certain progress. In the early studies, less polar solvents and inorganic ligands have been applied in the preparation process of CsPbBr3 QDs to decrease the density of oleic acid (OA) and oleylamine (OLA) [12, 13]. In order to avoid the loss of PLQY caused by the decrease of OA and OLA density during the purification process, short-chain organic ligands have been used to instead of long-chain organic ligands to modify the surface of AIP to increase the charge-transport property [14, 15]. In follow-up researches, the organic ligands with conjugated structure have been used to further increase charge-transport property of AIP via the intermolecular π-π interaction to avoid the insulation caused by aliphatic carbon chains [16, 17]. Compared with the phenethylammonium iodide, the aniline iodide without a carbon chain between the benzene ring and the substituent has shown more excellent modification property [18, 19], which indicates the substituent in conjugated organic ligands will also play an important role on the surface modification effect. Although a series of derivatives of s-triazine have shown outstanding charge-transport property in the application of perovskite solar cell [20, 21] and organic light-emitting diodes [22, 23], it is ignored based on them to modify AIP luminescent nanomaterials. Therefore, the surface modification strategy with s-triazine is expected to improve luminescence performance and charge-transport property of AIP simultaneously.
Here, s-triazine (TZ) and its derivatives (cyanuric acid named as CA and trithiocyanuric acid named as TA) were applied in surface modification strategy for enhancing photoelectric performance of AIP for the first time. The PLQY of AIP was increased to 88.15% and the current under 3V of the AIP film was enhanced from 4.44 mA to 81.20 mA with the modification of cyanuric acid (CA). Under the influence of hydroxyl group, CA has shown better modification ability than TZ. However, the effect of TA on the photoelectric performance of the AIP is negative for the sulfhydryl group will introduce extra free Pb2+ to cause more surface defects. The completely different surface modification results of CA and TA has shown the effectiveness of the improving the photoelectric performances and also indicated the potential risk of reducing PLQY of AIP either based on conjugated organic ligands at the meanwhile.
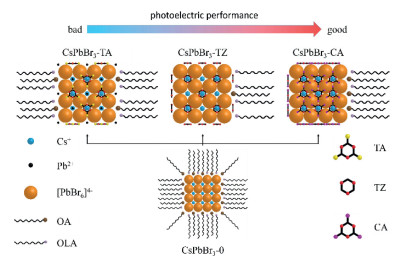
The AIP without the modification with 3N ligands was named as CsPbBr3-0. The AIP modified by TA, TZ and CA were named as CsPbBr3-TA, CsPbBr3-TZ and CsPbBr3-CA, respectively, the whole of which was named as CsPbBr3-3N. The CsPbBr3-0 and CsPbBr3-3N were are prepared with hot-injection method and the dosages of surface ligands were listed in Tables S1-S3 (Supporting information). The structures of AIP before and after modified with 3N ligands were shown in Fig. 1.

|
Download:
|
| Fig. 1. The structures of AIP before and after surface modification strategy: CsPbBr3-0, CsPbBr3-TA, CsPbBr3-TZ and CsPbBr3-CA. | |
{kind=link}
The solid powder X-ray diffraction (XRD) measurements were used to study the effect of three conjugated organic ligands on the host lattice of AIP. The obvious diffraction peaks can be found in the XRD patterns of the three organic ligands (Fig. S2 in Supporting information), indicating that AIP show goodish crystallization ability due to large polarity chemical bonds and the strong intermolecular π-π interaction happened in the 3N ligands. (Table S4 in Supporting information).
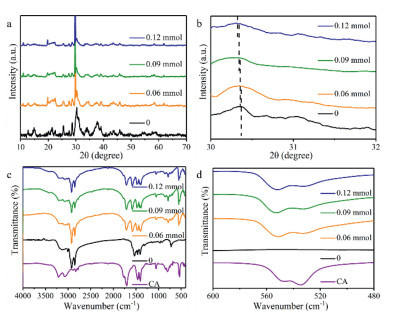
Before modified with 3N ligands, the characteristic diffraction peaks of CsPbBr3 around 15°, 22° and 30° can be observed in the XRD pattern of CsPbBr3-0, and the diffraction peaks between 30° and 31° are double peaks (Fig. S3 in Supporting information), which means the lattice structure of CsPbBr3-0 is orthogonal [24]. With the increasing the contents of TA, the double-peak diffraction peak around 30° moves to the low-angle region indicating that TA only induces the crystalline interplanar spacing to increase (Fig. S3). Compared with CsPbBr3-TA, the diffraction peaks located around 30° move to the low-angle region with a smaller amplitude in the XRD patterns of CsPbBr3-TZ (Fig. S4 in Supporting information). Especially when the dosage of TZ is increased to more than 0.09 mmol, the double diffraction peaks around 30° gradually become a single diffraction peak, indicating that TZ will induce AIP to form cubic crystal lattice (Fig. S4) [25]. The diffraction peaks of CA can be observed in the XRD patterns of CsPbBr3-CA (Figs. 2a and b), which indicates that CA will form crystalline layer on the crystal surface because of intermolecular hydrogen bonds and stronger intermolecular π-π interaction according to the theoretical calculation results (Table S4) [26]. Correspondingly, the position change of the peak around 30° in the XRD patterns of CsPbBr3-CA is the smallest among the three CsPbBr3-3N (Figs. 2a and b). When modified with 0.06 mmol CA, the diffraction peak around 30° becomes a single peak (Figs. 2a and b), meaning that CsPbBr3-CA has cubic crystal lattice. Thus, different substituents make these ligands have different influence on the host lattice.

|
Download:
|
| Fig. 2. (a) Full and (b) enlarged (from 30° to 32°) solid XRD patterns of CsPbBr3-0 and CsPbBr3-CA. (c) Full and (d) enlarged (from 480 cm−1 to 600 cm−1) FT-IR spectra of CsPbBr3-0 and CsPbBr3-CA. | |
{kind=link}
The compositions of the organic ligands in CsPbBr3-3N were analyzed on the basis of fourier transform infrared spectroscopy (FT-IR) measurements. The characteristic peak of C-S bond at 1120 cm−1 can be observed in the FT-IR spectrum of TA, which also can be found in that of CsPbBr3-TA (Fig. S5 in Supporting information). Moreover, there is a new peak at 1220 cm−1 in the FT-IR spectra of CsPbBr3-TA (Fig. S5). The intensity of the peak at 1220 cm−1 increases as the amount of TA increases, while the intensity of the peak at 1120 cm−1 decreases (Fig. S5), suggesting that the C-S bond in TA changes during the modification process with TA. In the FT-IR spectra of CsPbBr3-TZ, the characteristic peak of TZ located in the fingerprint region of 520 cm−1 can be directly observed, which moves to a low wavenumber region, indicating that the vibration of the chemical bond is limited after TZ adsorbing on the host lattice (Fig. S6 in Supporting information). Due to the tautomer of CA, there are characteristic peaks of hydroxyl group and carbonyl group in the FT-IR spectrum of CA, which are also found in the FT-IR spectra of CsPbBr3-CA without any change (Fig. 2c). The characteristic peak of CA located in the fingerprint area appears in the FT-IR spectra of CsPbBr3-CA and changes to a certain extent (Figs. 2c and d), meaning that CA interacts with the host lattice through the triazine ring instead of hydroxyl group or carbonyl group.
1H nuclear magnetic resonance (1H NMR) measurements were performed on CsPbBr3-0 and CsPbBr3-3N to obtain the chemical environment of the protons in these materials (Fig. S7 in Supporting information). The chemical environment of the proton hydrogen in CsPbBr3-TA is as same as CsPbBr3-0 (Fig. S7), indicating that the sulfhydryl group in TA will lose protons during the reaction, which changes the vibration state of the C-S bond. Compared with CsPbBr3-0, the proton signal of TZ can be clearly found in the 1H NMR spectrum of CsPbBr3-TZ (Fig. S7), which further suggests TZ have adsorbed on the host lattice. Meanwhile, the proton signals of the tautomers of CA in Fig. S7 suggest CA adsorb on the host lattice by triazine ring which is consisted with the analysis of FT-IR.
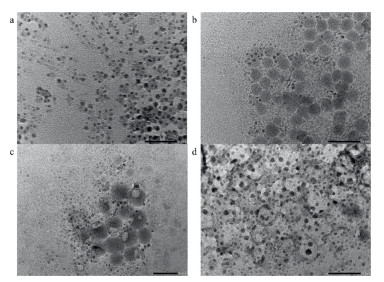
The effect of 3N ligands on the nanotopography of AIP was studied through transmission electron microscope (TEM) images. Before the modification with 3N ligands, CsPbBr3-0 shows various nanotopography in the TEM image, such as nanowires and nanocubes (Fig. 3a). Many regular nanospheres can be found in Figs. 3b and c, showing TA and TZ are beneficial to form the uniform nanotopography. The nanotopography of CsPbBr3-CA appears as a large cross-linked network (Fig. 3d), due to the crystalline layer on the surface of the host lattice. Therefore, CA exhibit significantly different influence on the nanotopography due to hydroxyl group.

|
Download:
|
| Fig. 3. TEM images (scale bar is 50 nm) of (a) CsPbBr3-0, (b) CsPbBr3-TA 0.09 mmol, (c) CsPbBr3-TZ 0.09 mmol and (d) CsPbBr3-CA 0.09 mmol. | |
{kind=link}
The elemental composition on the surface of the AIP was obtained by energy dispersive spectroscopy (EDS) measurements. As the amount of TA increases, the ratio of Br: Pb decreased from 3.26 to 3.07, 2.84 and 1.37 (Table S6 in Supporting information). The proton-losing sulfhydryl groups of TA will adsorb a large amount of free Pb2+ [27], which will increase surface defects of AIP to reduce PLQY [28]. The ratios of Br: Pb on the surface of CsPbBr3-TZ and CsPbBr3-CA are increased to 3.95, 4.27, 4.25, 4.09, 3.98 and 4.14, respectively (Tables S7 and S8 in Supporting information), which will enhance PLQY of AIP by decreasing surface defects [29]. Therefore, the surface structure and defect states of AIP have shown obviously opposite modification results under the influence of the 3N ligands with different substituents.
The absorption and photoluminescence (PL) spectra of AIP show different changes due to the different substituents on triazine ring. In the absorption spectra, maximum absorption wavelength of TA is the largest and that of CA is the smallest (Fig. S8 in Supporting information), which is consistent with the results of theoretical calculation (Table S5 in Supporting information). When the dosage of TA increases to 0.09 and 0.12 mmol, an absorption peak of 315 nm appears in the absorption spectra of CsPbBr3-TA, which is also present in the absorption spectra of CsPbBr3-TZ and CsPbBr3-CA, indicating that triazine ring will induce quasi-2D structure in the AIP (Fig. S8) [30]. It is worth noting that the absorption peak at 315 nm is unconspicuous in the absorption spectra of CsPbBr3-TA 0.09 mmol and CsPbBr3-TA 0.12 mmol, which is even barely visible in the absorption curve of CsPbBr3-TA 0.06 mmol, indicating that the modification ability of the triazine ring in TA is weakened due to the sulfydryl group (Fig. S8a). The emission peak of CsPbBr3-TA occurs blue-shift with the increase of ligand content, while the emission peaks of CsPbBr3-TZ and CsPbBr3-CA show red-shift first and then blue-shift (Figs. S9a, c, e in Supporting information).
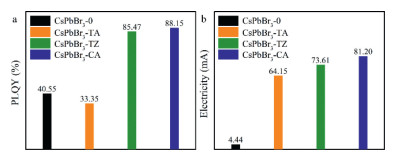
The PLQYs and fluorescence decay curves were obtained to study the effects of 3N ligands on the exciton recombination process of AIP (Figs. S9b, d, f in Supporting information), with which the radiation transition rate (kr) and the non-radiative transition rate (knr) of AIP were calculated. The knr of CsPbBr3-TA is larger than CsPbBr3-0 (Table S9 in Supporting information), indicating that the defect density on the surface of CsPbBr3-TA is greater than that of CsPbBr3-0, which is caused by excessive free Pb2+ on the surface. These changes result in that the PLQY of CsPbBr3-TA (1.24%-33.35%) is lower than CsPbBr3-0 (40.55%). When the amount of TA is 0.06 mmol and 0.12 mmol, the kr of CsPbBr3-TA is smaller than that of CsPbBr3-0, while the kr of CsPbBr3-TA 0.09 mmol is larger than that of CsPbBr3-0 (Table S9), for which the PLQY of CsPbBr3-TA 0.09 mmol (33.35%) is the highest among CsPbBr3-TA. The knr of CsPbBr3-TZ is smaller than CsPbBr3-0 (Table S10), indicating that TZ can well reduce the defect density on the surface of AIP. When the amount of TZ increases to 0.12 mmol, knr of the corresponding material began to increase (Table S10 in Supporting information), indicating that the excessive TZ at this time causes the interplanar spacing of the host lattice to change too much, which would increase the defect density on the surface. Similar to CsPbBr3-TA, the kr of CsPbBr3-TZ shows a trend of first decreasing, then increasing and then decreasing with the increase of TZ dosage (Table S10). As for CsPbBr3-TZ 0.09 mmol, the kr reaches the maximum and knr is at the minimum, achieving the highest PLQY among CsPbBr3-TZ (85.47%). After modifying the host lattice with CA, the knr of AIP decreases with the amount of CA increasing (Table S11 in Supporting information), which means that CA can significantly reduce the surface defect density of AIP. Different from CsPbBr3-TA and CsPbBr3-TZ, the knr of CsPbBr3-CA 0.12 mmol is less than CsPbBr3-CA 0.09 mmol (Table S11). Combined with the XRD analysis results, CA has little effect on the interplanar spacing of the host lattice, which can be inferred that 0.12 mmol of CA will not cause a large interplanar change and increase surface defects. The kr of CsPbBr3-CA shows the same trend as CsPbBr3-TA and CsPbBr3-TZ (Table S11). Correspondingly, CsPbBr3-CA 0.09 mmol exhibits the largest PLQY (88.15%). Since the three organic ligands all contain triazine ring, it can be inferred that an appropriate amount of triazine ring can effectively increase the kr of AIP. These three triazine ring-based organic ligands exhibit different influence on PLQY of AIP due to the different substituents (Fig. 4a).

|
Download:
|
| Fig. 4. (a) PLQY of CsPbBr3-0 and CsPbBr3-3N 0.09 mmol solid powder (λex = 390 nm). (b) Electricity of CsPbBr3-0 and CsPbBr3-3N 0.09 mmol films under 3 V. | |
{kind=link}
Since the kr of CsPbBr3-TA 0.09 mmol, CsPbBr3-TZ 0.09 mmol, and CsPbBr3-CA 0.09 mmol are all higher than CsPbBr3-0, variable temperature fluorescence measurements were performed on these four materials to further explore the reasons. In Fig. S10 (Supporting information), CsPbBr3-0 shows four emission peaks at low temperature, while CsPbBr3-TA 0.09 mmol, CsPbBr3-TZ 0.09 mmol and CsPbBr3-CA 0.09 mmol all maintained single emission peak (Fig. S10), indicating that the nanotopography of CsPbBr3-3N is more uniform, which coincides with the TEM images. In Fig. S10, the emission intensity of CsPbBr3-0 begins to decrease rapidly at 115 K as the temperature rises, while this temperature is increased to 230 K (CsPbBr3-TA 0.09 mmol), 150 K (CsPbBr3-TZ 0.09 mmol) and 150 K (CsPbBr3-CA 0.09 mmol) after the surface modification with 3N ligands. This result means that all of the 3N ligands can increase the exciton binding energy of AIP to increase the kr of AIPs [31]. Although CsPbBr3-TA 0.09 mmol has shown the largest exciton binding energy, due to the influence of surface defects caused by sulfhydryl group, the kr of CsPbBr3-TA 0.09 mmol is smaller than that of CsPbBr3-CA 0.09 mmol. Therefore, the surface state of the AIP can be tuned through controlling the amount of ligand and the type of substituent, so as to realize the optimization on the luminescence performance of AIP.
In the discussion about luminescence performance, CsPbBr3-TA 0.09 mmol, CsPbBr3-TZ 0.09 mmol and CsPbBr3-CA 0.09 mmol have shown better luminescence performance (Fig. 4a). Therefore, the content of OA and OLA in these three materials and CsPbBr3-0 and the charge-transport property of the material films were tested respectively. The contents of organic ligands in these four materials were obtained through DTG measurements (Table S12 in Supporting information). The mass percentage of OA and OLA in the CsPbBr3-0 totals 27.7%, which is significantly reduced in CsPbBr3-3N, suggesting that the 3N ligands can significantly reduce the density of OA and OLA on the surface of AIP. The charge-transport property of these four kinds of AIP films were measured through a device with a structure of ITO/AIP/Al. The current-voltage curve in the film under the forward voltage is shown in Fig. S11 (Supporting information). The currents in CsPbBr3-0, CsPbBr3-TA 0.09 mmol, CsPbBr3-TZ 0.09 mmol and CsPbBr3-CA 0.09 mmol films gradually increase under the same voltage (Fig. 4b), which proves that the organic ligands containing the triazine ring exhibit two notable advantages for the great enhancement on the charge-transport property of AIP: strong intermolecular π-π interaction and lowering the density of OA and OLA on the surface of AIP [32]. Among the CsPbBr3-3N films, CsPbBr3-TA 0.09 mmol shows the worst charge-transport property, because the more surface defects caused by TA will reduce the charge-transport property of AIP [33]. The CsPbBr3-CA 0.09 mmol shows the best charge-transport property due to the stronger modification ability and intermolecular π-π interaction of CA inferred from the results of theoretical calculation. Therefore, 3N ligands can greatly improve the charge-transport property of AIP, and CA shows the best the optimization effect.
We successfully improve the photoelectric performance of AIP via changing the substituents to optimize the modification ability of conjugated organic ligand. According to the results of XRD, FT-IR, PL and other measurements, it can be proved that the surface modification strategy with s-triazine can effectively improve the luminescence performance and charge transport performance of AIP through the triazine ring structure. The addition of appropriate groups (such as hydroxyl) to the triazine ring structure can significantly enhance the modification effect of organic ligands. Meanwhile, the inappropriate substituents (such as sulfhydry) will introduce additional surface defects on the surface of AIP and reduce the modification effect of organic ligands. This study leads researchers to notice the great influence of molecular design strategy on the modification effect of organic ligand in surface modification strategy of AIP.
Declaration of competing interestThe authors declare no conflict of interest.
AcknowledgmentsThis work was funded by National Natural Science Foundation of China (No. 52073045), the Key Scientific and Technological Project of Jilin Province (No. 20190701010GH), and the Development and Reform Commission of Jilin Province (No. 2020C035-5). D. Zhu is grateful for the support from the Key Laboratory of Nanobiosensing and Nanobioanalysis at the Universities of Jilin Province. The authors acknowledge the support from the Jilin Provincial Department of Education.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.06.066.
| [1] |
L. Protesescu, S. Yakunin, M.I. Bodnarchuk, et al., Nano Lett. 15 (2015) 3692-3696. DOI:10.1021/nl5048779 |
| [2] |
G.L. Yang, H.Z. Zhong, Chin. Chem. Lett. 27 (2016) 1124-1130. DOI:10.1016/j.cclet.2016.06.047 |
| [3] |
L. Cheng, Y. Cao, R. Ge, et al., Chin. Chem. Lett. 28 (2017) 29-31. DOI:10.1016/j.cclet.2016.07.001 |
| [4] |
M. Imran, V. Caligiuri, M. Wang, et al., J. Am. Chem. Soc. 140 (2018) 2656-2664. DOI:10.1021/jacs.7b13477 |
| [5] |
M. Lu, J. Guo, S. Sun, et al., Chem. Eng. J. 404 (2021) 126563-126569. DOI:10.1016/j.cej.2020.126563 |
| [6] |
N. Mondal, A. De, A. Samanta, ACS Energy Lett. 4 (2019) 32-39. DOI:10.1021/acsenergylett.8b01909 |
| [7] |
Y. Nanishi, Nat. Photon. 8 (2014) 884-886. DOI:10.1038/nphoton.2014.291 |
| [8] |
K.H. Kim, J.J. Kim, Adv. Mater. 30 (2018) 1705600-1705618. DOI:10.1002/adma.201705600 |
| [9] |
J. Dai, J. Xi, Y. Zu, et al., Nano Energy 70 (2020) 104467-104473. DOI:10.1016/j.nanoen.2020.104467 |
| [10] |
O. Elimelech, O. Aviv, M. Oded, U. Banin, Nano Lett. 20 (2020) 6396-6403. DOI:10.1021/acs.nanolett.0c01913 |
| [11] |
A. Loiudice, M. Strach, S. Saris, D. Chernyshov, R. Buonsanti, J. Am. Chem. Soc. 141 (2019) 8254-8263. DOI:10.1021/jacs.9b02061 |
| [12] |
N.K. Kumawat, A. Swarnkar, A. Nag, D. Kabra, J. Phys. Chem. C 122 (2018) 13767-13773. DOI:10.1021/acs.jpcc.8b00723 |
| [13] |
B.B. Zhang, S. Yuan, J.P. Ma, et al., J. Am. Chem. Soc. 141 (2019) 15423-15432. DOI:10.1021/jacs.9b08140 |
| [14] |
Y. Zhu, J. Zhao, G. Yang, X. Xu, G. Pan, Nanoscale 12 (2020) 7712-7719. DOI:10.1039/d0nr01378a |
| [15] |
J.H. Park, A.Y. Lee, J.C. Yu, et al., ACS Appl. Mater. Interfaces 11 (2019) 8428-8435. DOI:10.1021/acsami.8b20808 |
| [16] |
Y.K. Wang, Z.Q. Jiang, L.S. Liao, Chin. Chem. Lett. 27 (2016) 1293-1303. DOI:10.15244/pjoes/61701 |
| [17] |
H. Shao, Y. Zhai, X. Wu, et al., Nanoscale 12 (2020) 11728-11734. DOI:10.1039/d0nr02597f |
| [18] |
G. Li, J. Huang, H. Zhu, et al., Chem. Mater. 30 (2018) 6099-6107. DOI:10.1021/acs.chemmater.8b02544 |
| [19] |
T. Chiba, Y. Hayashi, H. Ebe, et al., Nat. Photon. 12 (2018) 681-687. DOI:10.1038/s41566-018-0260-y |
| [20] |
F. Yang, M.A. Kamarudin, D. Hirotani, et al., Sol. RRL 3 (2019) 1800275-1800280. DOI:10.1002/solr.201800275 |
| [21] |
J. Liu, D. Wang, K. Chen, et al., Solar Energy 206 (2020) 548-554. DOI:10.1364/oe.381503 |
| [22] |
R.A. Klenkler, H. Aziz, A. Tran, Z.D. Popovic, G. Xu, Org. Electron. 9 (2008) 285-290. DOI:10.1016/j.orgel.2007.11.004 |
| [23] |
M.A. Fusella, R. Saramak, R. Bushati, et al., Nature 585 (2020) 379-382. DOI:10.1038/s41586-020-2684-z |
| [24] |
Y. Wang, F. Yang, X. Li, et al., Adv. Funct. Mater. 29 (2019) 1904913-1904921. DOI:10.1002/adfm.201904913 |
| [25] |
S. Wang, C. Bi, J. Yuan, L. Zhang, J. Tian, ACS Energy Lett. 3 (2018) 245-251. DOI:10.1021/acsenergylett.7b01243 |
| [26] |
M.N. An, S. Park, R. Brescia, et al., ACS Energy Lett. 6 (2021) 900-907. DOI:10.1021/acsenergylett.1c00052 |
| [27] |
Y. Hassan, J.H. Park, M.L. Crawford, et al., Nature 591 (2021) 72-77. DOI:10.1038/s41586-021-03217-8 |
| [28] |
F. Li, Y. Liu, H. Wang, et al., Chem. Mater. 30 (2018) 8546-8554. DOI:10.1021/acs.chemmater.8b03442 |
| [29] |
Y. Zu, J. Dai, L. Li, et al., J. Mater. Chem. A 7 (2019) 26116-26122. DOI:10.1039/c9ta08421e |
| [30] |
J. Lu, Z. Wei, J. Semicond. 41 (2020) 051203-051210. DOI:10.1088/1674-4926/41/5/051203 |
| [31] |
K. Wu, A. Bera, C. Ma, et al., Phys. Chem. Chem. Phys. 16 (2014) 22476-22481. |
| [32] |
Y.H. Suh, T. Kim, J.W. Choi, C.L. Lee, J. Park, ACS Appl, ACS Appl. Nano Mater. 1 (2018) 488-496. DOI:10.1021/acsanm.7b00212 |
| [33] |
C. Qin, A.S.D. Sandanayaka, C. Zhao, et al., Nature 585 (2020) 53-57. DOI:10.1038/s41586-020-2621-1 |