 2022, Vol. 33
2022, Vol. 33 


b Institute of Environmental and Energy Catalysis, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China;
c College of Resources and Environmental Engineering, Tianshui Normal University, Tianshui 741001, China;
d School of Sciences, Xi'an Technological University, Xi'an 710021, China
Ammonia (NH3), as a future carrier of renewable energy and a source of fertilizer in agriculture, is the key aspects and anticipated stages in promising technologies for decades, which is provided with large hydrogen content and high energy density [1-3]. Although electrosynthesis of NH3 from N2 composing of 78% of the atmosphere, which is a fertile source for synthetic ammonia, it exists in chemically and biologically unusable gaseous form. Haber, who break through the triple bond of dinitrogen with hydrogen in the presence of the Fe and discovered the process by high temperatures and pressures, explained that his main motivation for the study of synthesizing ammonia was the growing demand for food, and is awarded a Nobel Prize in 1931 [4]. Recently, more and more researchers are paying attention to the approach to replace the Haber-Bosch process by searching for the befitting catalyst support of ceria under ambient conditions. Doping single metals of chromium (Cr), copper (Cu), iron (Fe), molybdenum (Mo) and rubidium (Ru) can enhance the performance of electrocatalytic N2 to NH3 in comparison to the pure CeO2(111) surfaces efficiently, which the single Mo atoms in Mo-N3C are also reported for the reason of the best capability of N2 adsorption for further electrochemical N2 reduction [5-9]. Lee and co-workers explored the new way called the reticular chemistry approach which exploiting MOFs water-repelling and molecular-concentrating effects to overcome HER-imposed bottlenecks to accelerate synthesis yield of ammonia in theory [10]. Also, Guo et al. focus on the atomically dispersed Bi-catalysts for nitrogen reduction reaction to tackle the activity and selectivity [11]. As well as Mxene-based materials, which have been noticed as highlighted catalysts for electrochemical N2 reduction recently, are investigated rapidly due to satisfactory catalytic activity [12]. Analogously, the process of Ammonia borane (AB) hydrolysis generates H2 and NH3 on the transition metal Fe@Co core-shell structure has been obtained [13, 14].
However, we will have to admit and should not ignore that the difficulty of a strong N-N bond is the biggest problem in the synthesis of ammonia. In a word, it makes us provide the ideas for seeking another supplementary scheme. As a nitrogen supplement, nitric oxide (NO) has the advantage of bond energy over nitrogen molecules, which consumes much less energy to synthesize ammonia. Especially on the surfaces of CeO2 (111), oxygen vacancies play an important role in good catalytic performance, which comes from the interconversion between Ce(III) and Ce(IV) oxidation states with the storage and release of 4f electronic orbits [14-19]. In this work, our study demonstrates that we built a 3 × 3 × 1 supercell of CeO2 (111) surface by doping La atom with oxygen vacancy for adsorbing NO molecules to study geometry, electronic structures, and NO reduction reaction coordinate to propose a new idea of N-element reduction reaction (NORR):
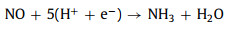
|
(1) |
The calculations were performed by a spin-polarized DFT + U approach using the Vienna ad initio simulation package (VASP) [20], which employing the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerh of (PBE) function. Also, we use DFT-D2 to describe the van der Waals bonds. For guaranteeing a good convergence of total energies, the plane-wave cutoff was 400 eV, which uses the Brillouin zone sampled with 5 × 5 × 1 of k-points with allowing the convergence of total energy to set 0.01 eV, and the Part valence-electron configurations include Ce (5s, 5p, 6p, 5d, 4f), La (5d, 6s) and O (2s, 2p). And we consider the 4f states of the reduced cerium and doped-La atoms, where the value of the Hubbard U terms was used as 5.0 eV and 7.5 eV effectively [21], and we first predicted bulk lattice constant of the pure CeO2 is 5.42 Å, which compared it and the agreement with the experimental value (5.41 Å) and the theoretical results (5.43 Å) [22, 23]. For the surface selection, we studied only the surface (111) among the low-index surfaces of CeO2 (111), (110) and (100), which surface (111) is the most stable [24, 25]. As depicted in Fig. S1 (Supporting information), the circular area of the dotted line represents the possible NO adsorption sites of Ce-top, O-top, Ce-O-bridge and hollow sites.
For purpose of better understanding the transition metal (TMs) doped CeO2 (111) surfaces the formation energies of each surface slab by using the following equation:
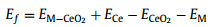
|
(2) |
where EM−CeO2 and ECeO2 are total energies of TMs doped and stoichiometric CeO2 (111) surface, respectively. ECe and EM are the energies of single Ce atom and the selected metal atom (M = K, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Mo, Cs, Ba, La, Pr, Nd and Sm), respectively. Clearly, the negative value presents the exothermic process, leading to the stable structure. When the values were calculated to be positive, it explained that doping process is thermodynamically unfavorable.
As shown in Fig. S2 (Supporting information), all the formation energies of the metal-doped CeO2 (111) surfaces are plotted, we actually see that except Cs elements, the dopant formation energies of K to Ba atoms (> 2 eV) are higher than those (< 1.5 eV) which doped with rare metals (La, Pr, Nd and Sm). To further compare with rare metals, La-doped CeO2 (111) surface with the lowest formation energy of 0.67 eV indicating that the syntheses of La-doped-CeO2 (111) systems are relatively easy in the experiment. Our results are completely consistent with the radii between the M and Ce atoms, that the larger the size difference is, the larger the formation energy is [26].
Generally, oxygen vacancy (OV) plays an important role in good catalytic performance, which coming from the interconversion between Ce(III) and Ce(IV) oxidation states with the storage and release of 4f electronic orbits. Thus, for investigating the formation energy of oxygen vacancies (OVs) on the CeO2 (111) surface, the defect energy Evac is defined as following [27, 28]:
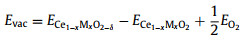
|
(3) |
where ECe1−xMxO2−δ, EO2 and ECe1−xMxO2 are the energy of the bulk in the presence of one oxygen vacancy, the energy of a gas-phase O2 and the bulk of surface Ce1-xMxO2, respectively. The definition shows that a positive value indicates an endothermic-formation and a negative value indicates an exothermic formation. We use the same level to gain the formation energy of OVs in this work. As shown in Fig. S1, the formation energy of an oxygen vacancy on the TMs and rare metals doped surface is lower value, compared with the pure surface of 3.01 and 2.85 eV [29, 30], and this fully demonstrated the doping ways can promote the formation of oxygen vacancies, which are in agreement with the calculation by Xue et al. [31, 32].
The adsorption energy (Eads) is defined as [33, 36]:

|
(4) |
where Eadsorbate/sub, Eadsorbate and Esub are the total energies of adsorbate-substrate, isolated adsorbate and substrate system, respectively. And the negative value of Eads indicates the better stable configuration and exothermic process.
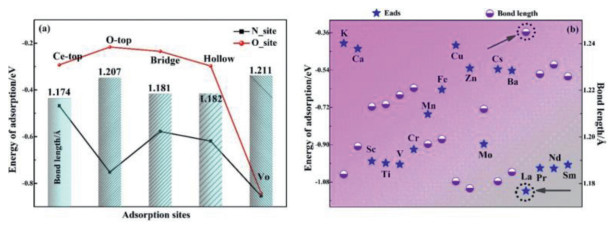
As shown in Fig. 1a and Fig. S1, in order to further elucidate and study the nature of the CeO2 systems for NO activation, we have tested Eads of and bond length of NO, the x-axis coordinates indicate the possible adsorption locations, which are Ce-top, O-top, bridge, hollow and OV sites and the y-axis is the energy of adsorption. Also, we can see that red and black lines, which show NO molecule can be adsorbed over different sites through its O and N atom, respectively. The bar chart shows the bond length of NO after adsorption through N atom, and the date by O atom is listed in Table S1 (Supporting information) due to the minor change. Our computations demonstrated that NO chemisorption occurs on the N site preferably, which the value of N-site shows better adsorption than O-site with Eads under −0.40 eV in general. Moreover, when the OV occurs on the surface, the adsorption energy is significantly reduced, and the bond length is also increased of 1.211 Å. In all, the results indicate that the existence of OV can well improve the ability of NO activation. Especially, adsorption energy and bond length are significantly improved when metals are doped with oxygen vacancy. By comparison of different adsorption energy Eads of NO and bond length after different metals doping on MxCe1-xO2-δ surfaces (M = K, Ca, Sc, Ti, V, Cr, Mn, Fe, Cu, Zn, Mo, Cs, Ba, La, Pr, Nd and Sm), what is noteworthy is that the bond length of NO molecule increased to 1.245 Å from its original length of 1.117 Å and the adsorption energy decreased to −1.12 eV by activating over La-doping with OV surface (Fig. 1b).

|
Download:
|
| Fig. 1. (a) The adsorption energy of NO (Eads) for different sites and corresponding bond length on the clean CeO2 surface and CeO2- urface with an oxygen vacancy, as well as the calculation of the value on the MCe1-O2- surfaces (M = K, Ca, Sc, Ti, V, Cr, Mn, Fe, Cu, Zn, Mo, Cs, Ba, La, Pr, Nd and Sm). Thus the results for Co and Ni doping are not shown in the work because of nonconvergent. | |
{kind=link}
In a word, according to Fig. 1, Tables S1 and S2 (Supporting information), we found that direct generation of OV can also achieve the result of NO activation (the formation energy of oxygen vacancy (Eads) is 3.01 eV, the bond length of NO is 1.211 Å and the Eads is –0.85 eV), except the system of low formation energy doping La atom with OV (the formation energy of oxygen vacancy (Eads) is 2.85 eV, the bond length of NO is 1.245 Å and the Eads is –1.12 eV). As shown in Fig. S4 (Supporting information), NO molecule prefers to adopt an end-on way with Ce-N bonding distance of 2.491 Å on the OV surface and a side-on mode with Ce-O, Ce-N and La-N bonding distances of 2.703, 2.542 and 2.661 Å. It is obvious that the defect area is large, the distance of Ce3+-O is too far to bond on pure OV surface. Conversely, duo to the La ion is bigger that the Ce and the defect area is smaller, N and O atom can contact with more reductive active sites at the same time to pull the NO bond. Therefore, in order to further study the difference between them for NORR, we carried out the HER study over La doped CeO2 (111) surface with an oxygen vacancy and CeO2 (111) surface only with OV, respectively (Fig. S5 in Supporting information), which is an important side reaction to hinder the process of NORR. Ulteriorly, we can see that the first hydrogen is adsorbed on the La doped CeO2(111) surface with an oxygen vacancy and CeO2(111) surface only with Vo with the free energy change of −0.73 and −0.81 eV, respectively, which the calculated equation is ΔGH = Eads + 0.24 [36]. In other words, the results reveal the practical free energies change of *H are −0.97 eV over La-doped CeO2 with Vo, which is smaller than that of *NO (−1.12 eV) and the Eads of *H are −1.05 eV over pure CeO2 with Vo, which is bigger than that of *NO (−0.85 eV). The calculation indicates that the La-doped CeO2 (111) with OV can dominantly attract more NO than H, where the defected surface can expose more active sites for NORR.
To speculate the mechanism of the adsorption sites of the NO-molecule, it is necessary to calculate the difference charge density after doping La atom and forming an oxygen vacancy. We thus investigate the electronic structures of interaction between NO and defected species and densities of states (DOS) of N and O atoms which guiding us to determine the location of the reduction sites [37]. According to the Fig. 2a, the charge transference from OV site to La2+ (Bader charge of 1.71|e|) and Ce3+ (Bader charge of 2.81|e|) sites, respectively. Now therefore, the existence of chemical adsorptive sites may at the position of Ce3+ and La2+.

|
Download:
|
| Fig. 2. (a) Difference charge density of CeO2 (111) surface with defect, and red color (0.003 eV/Å3) in the plot indicates electron density increase after formation, and blue represents the opposite. (b) The calculated geometries of dCe-O, dLa-O and dCe−M (M = Ce and La) are shown to compare with pure CeO2 (111) surfaces. (c) The spin-polarized density of states (DOS) of the optimized stoichiometric ceria. The fermi level is set at 0 eV. | |
{kind=link}
For defected systems containing a single metal atom doping and an oxygen vacancy, only the lanthanide element is used, which the value of Eads is lowest and degree of NO bond activation shows the best performance of 1.245 Å, giving smaller to the volume of dopant hole, where is enclosed by i, ii and iii in Fig. S1, which considering the reason of the similar ion radius sizes between Ce and La atom. Thus, the combination of Ce3+ and La2+ active reduced sites together can accelerate NO molecules activation, were reacting on a finite scale. As shown in Fig. 2b, the red and blue patterns indicate the distances of neighboring atoms each other on the pure surface and defective surfaces, respectively. The distances between M and Ox (x = I, II and III) atoms (M = Ce and La) are 2.371, 2.369, 2.339 and 2.241 Å as shown in Fig. S1 and Fig. 2b, which are closed to the distance of pure surface (2.348 Å) [38-40]. To better describe the situation of the active sites, it is worth mentioning that the density of states (DOS) to study the influence of doping La atom with an oxygen vacancy on the electronic structure of defected CeO2 surface. Fig. 2c shows that the orbital electronics of Ce(f) contributed between −0.2 eV and 0 eV, and La(d) orbital electronics contributed between −1.85 eV and 0 eV apparently with both VB occupied by states in spin up closed to the Fermi level, that the result is well consistent with the value [41].
For further understanding activation of NO molecule, we make the Bader charge analysis that N atom accepts the charge of 0.53|e| from the Ce and La cation and O atom receives the charge of 0.17|e| (Fig. 2b). The evidence of chemical adsorption can be confirmed by the charge density difference and is calculated using the expression [42-46]:
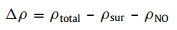
|
(5) |
where ρtotal, ρsur and ρNO are the charge densities of the NO adsorption on the OV site with metal-La doping, the charge density of the pure surface, and the charge density of studied gas molecule-NO, respectively [33].
The Gibbs free energy change (ΔG) for each elementary step was obtained using the computational hydrogen electrode (CHE) model by the following equation [45]:

|
(6) |
where ΔE, ΔEZPE and ΔS are the reaction energy by the DFT calculation and the changes in zero-point energy and entropy, respectively. T is the temperature (298.15 K).
In a word, the adsorption energy of NO on the OV site indicates that the CeO2 (111) surface is modified by metal La and vacancy of O atom with a synergistic effect. Charge density difference plots (Figs. 3a and b) show that adsorbed NO can interact with these active and occupied orbitals of La and Ce atoms, in which the lone pair electrons from the Ce3+ and La2+ are injecting into the electrons from the Ce3+ and La2+ are injecting into the antibonding orbitals of NO via N and O atoms by the so-called as the "push" hypothesis. The more interaction is thus for the lower dissociation barrier than NO adsorption on the pure surface. Our calculation is further performed to provide more details of such chemical change between isolated La and NO by fragment orbital analysis as shown in Figs. 3c–g [11, 46-52]. Also, the projected densities of states (PDOS) of NO adsorption on defected species show that α-, β- and αβ-spin states represent pure substrate surface, a gaseous NO molecule, and the adsorption configuration for comparison (Figs. 3f and g) [53].

|
Download:
|
| Fig. 3. All calculation for electronic structure analysis. (a, b) Optimized structure of adsorption configurations and charge density differences about NO chemisorbed on the CeO2 (111) partial slab models from the top views. The isosurfaces value is 0.003 eV/Å3. (c–e) The simplified schematic with bonding in NO molecule and metal atoms (La and Ce) on the defective surfaces. (f, g) The projected electronic of states (PDOS) with spin-polarized density. The fermi level is set at 0 eV. | |
{kind=link}
For the interaction between the two main states of 4f-5d for Ce3+ atom and O2p states for O atoms, we present in Figs. 3c, e and g that the energy levels of Ce3+ majority f spin states involved in the interaction with p-spin orbitals of O atom with the energy of αβ-states lower which becoming more stable. Obviously, the fx3 orbitals electrons are donated to the O2p orbitals, which proving up to the hilt that surface generated an OV possess the capability of redox. Also, for interaction between Ce3+, La2+ and O atoms, we present in Figs. 3d–f the spin densities are strongly localized on Ce4f, 5d, La5d and 2p of N states, the energy levels of Ce3+ majority 4f spin states and La2+ 5d-states involved in the interaction with 2p-spin orbitals of N atom with the energy of αβ-states lower which becoming more stable, too. Similarly, as our previous analysis, the 4fx3 orbitals of Ce and 5dz3 orbitals of La electrons are donated to the 2p orbitals of N indicates that surface-anchored La also owns the capability of redox.
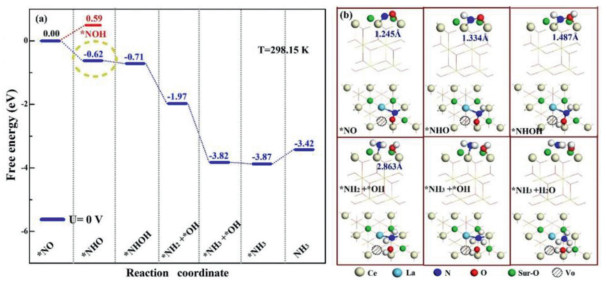
Critically, the investigation into the mechanism of ammonia synthesis over the defected surface with the raw material (NO) is essential. Hence, to explore the reaction path of NORR on La-doped CeO2 catalysts, the associative hydrogenation of pathways, which the proton-electron pair (H+ + e−) attacks the N atom firstly and then O atom continuously until the NH3 is generated, was considered according to the analysis of the degree of activation mechanism between N and O atom and the horizontal adsorption configuration, thus the barrier of NO direct dissociation greater than associative hydrogenation. And the Gibbs free energy of each reaction intermediate was calculated as shown in Fig. 4a.

|
Download:
|
| Fig. 4. (a) Free energy diagram of the NORR on the surface of La-doped CeO2 (111) with OV, and all the calculations of free energy are at U = 0 V and T = 298.15 K. (b) The top and side views of optimized structures of NO adsorption on the defect CeO2(111) following the NORR pathways. | |
{kind=link}
Different from the value on the O site, ΔG(*NHO) of −0.62 eV is lower than the ΔG(*NOH) of 0.59 eV as shown in Fig. 4a which comparing with other theoretical calculation of positive value and showing the excellent performance [54, 55]. We further examine the "enzymatic" pathway that the applied potential for U = 0 V and T = 298.15 K to study the true condition [56, 57]. And the detailed thermodynamic date we calculated the Gibbs free energy of all the adsorption and desorption structures are considered as shown in Fig. 4b, which the length of NO bond changes of 1.244, 1.334, 1.487 and 2.863 Å in the process of hydrogenation until breaking. Remarkably, due to the degree of NO activation and its own activity, whose *NxOy (x = 1, 2 and 3; y = 1, 2) intermediate is substantially low, resulting in a smaller energy barrier of −0.62 eV for the first hydrogenation of *NO into *NHO, which is an exothermic reaction. For the second and third step of *NHOH and *NH2OH, the reactions are both exothermic energies of −0.09 and −0.26 eV to promote the dissociation of NO, that the third step (*NH3 + *OH with the NO bond splitting up) is also downhill. It is presented in Fig. 4a that Vo with La doping favors the *NH3 + *H2O stabilization with −1.85 eV downhill for *HH2 + *OH → *NH3 + *OH substantially. By the step of fifth for *NH3 + *H2O, we also found that NORR activity of La-CeO2-OV over surface (111) can construct H2O. For the last step, the reason that we ignored the consideration for NORR process is its form of NH4+. Thus, the NORR process we conclude is thermodynamically spontaneous.
Based on the theoretical findings, the NORR performance of La-CeO2-OV on the associative hydrogenation reaction coordinate can be described as illustrated in Fig. 4. Firstly, the direction of alternation is determined by the activation mechanisms between N and O atoms and the horizontal adsorption configuration. Second, the situations of La-dopants and OV will effectively push the orbitals electrons of the sites of Ce and La to the NO molecular orbitals, resulting in the accelerated transmission of proton-coupled electrons and the spontaneous reaction that synthesis of the ammonia and water.
Subsequently, we find that the effect of atomic radii is a critical factor for altering and ensuring the appropriate size of the NO molecule adsorption. We have also recorded that the most active reduction surface predicted is OV site, and investigated the CeO2(111) with doping the La metals from the ab initio DFT calculations by studying geometric and electronic structure properties. The results show that Eads is of −1.12 eV with relative horizontal adsorption, the Bader charge analysis of N atom accepting the charge of 0.53|e| from the Ce and La cation and O atom receiving the charge of 0.17|e|. Importantly, ΔG is less than zero of NORR (NO + 5(H+ + e−) → NH3 + H2O), which indicating thermodynamically spontaneous reaction to synthesize ammonia and water under ambient conditions.
In summary, we have demonstrated the good structural engineering of reduction surface by ceria when La-doping with Vo. Comparing with NRR coordinates, NORR has lower energy consumption as confirmed by the theoretical results [46]. And thus, we anticipate that such metals-La including oxygen vacancies can be extended over new ideas for efficient electrocatalytic NO to ammonia as a feedstock of nitrogen supplement.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis study was funded by the Natural Science Foundation of China (No. 21603109), the Henan Joint Fund of the National Natural Science Foundation of China (No. U1404216), the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0676) and the Special Fund of Tianshui Normal University, China (No. CXJ2020-08).
Appendix Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.05.072.
| [1] |
L.J. Arachchige, Y. Xu, Z. Dai, et al., J. Phys. Chem. C 124 (2020) 15295-15301. DOI:10.1021/acs.jpcc.0c03899 |
| [2] |
R. Wang, C. He, W. Chen, et al., Chin. Chem. Lett. (2021) 10.1016/j.cclet.2021.05.024. DOI:10.1016/j.cclet.2021.05.024 |
| [3] |
J. Wang, C. He, J. Huo, et al., Adv. Theory Simul. 4 (2021) 2100003. DOI:10.1002/adts.202100003 |
| [4] |
J.W. Erisman, M.A. Sutton, J. Galloway, et al., Nat. Geosci. 1 (2008) 636-639. DOI:10.1038/ngeo325 |
| [5] |
K. Chu, Y.H. Cheng, Q.Q. Li, et al., J. Mater. Chem. A 8 (2020) 5865-5873. DOI:10.1039/c9ta14260f |
| [6] |
H. Xie, H. Wang, Q. Geng, et al., Inorg. Chem. 58 (2019) 5423-5427. DOI:10.1021/acs.inorgchem.9b00622 |
| [7] |
S.B. Zhang, C.J. Zhao, Y.Y. Liu, et al., Chem. Commun. 55 (2019) 2952-2955. DOI:10.1039/c9cc00123a |
| [8] |
J. Qi, L. Gao, F. Wei, Q. Wan, S. Lin, ACS Appl. Mater. Interfaces 11 (2019) 47525-47534. DOI:10.1021/acsami.9b15570 |
| [9] |
L. Chen, C. He, R. Wang, et al., Chin. Chem. Lett. 32 (2021) 53-56. DOI:10.1016/j.cclet.2020.11.013 |
| [10] |
H.K. Lee, C.S.L. Koh, Y.H. Lee, et al., Sci. Adv. 4 (2018) eaar3208. DOI:10.1126/sciadv.aar3208 |
| [11] |
X. Guo, J. Gu, S. Lin, et al., J. Am. Chem. Soc. 142 (2020) 5709-5721. DOI:10.1021/jacs.9b13349 |
| [12] |
J. Sun, W. Kong, Z. Jin, et al., Chin. Chem. Lett. 31 (2020) 953-960. DOI:10.1016/j.cclet.2020.01.035 |
| [13] |
J. Huo, L. Fu, C. Zhao, C. He, Chin. Chem. Lett. 21 (2021) 2269-2273. DOI:10.1016/j.cclet.2020.12.059 |
| [14] |
J. Huo, J. Wang, H. Yang, et al., J. Mol. Model. 27 (2021) 38. DOI:10.1007/s00894-020-04628-6 |
| [15] |
T. Montini, M. Melchionna, M. Monai, P. Fornasiero, Chem. Rev. 116 (2016) 5987-6041. DOI:10.1021/acs.chemrev.5b00603 |
| [16] |
X. Liu, K. Zhou, L. Wang, B. Wang, Y. Li, J. Am. Chem. Soc. 131 (2009) 3140-3141. DOI:10.1021/ja808433d |
| [17] |
A. Trovarelli, J. Llorca, ACS Catal. 7 (2017) 4716-4735. DOI:10.1021/acscatal.7b01246 |
| [18] |
J. Paier, C. Penschke, J. Sauer, Chem. Rev. 113 (2013) 3949-3985. DOI:10.1021/cr3004949 |
| [19] |
L. Liu, A. Corma, Chem. Rev. 118 (2018) 4981-5079. DOI:10.1021/acs.chemrev.7b00776 |
| [20] |
M.C. Wasson, X. Zhang, K.I. Otake, Chem. Mater. 32 (2020) 8522-8529. DOI:10.1021/acs.chemmater.0c02740 |
| [21] |
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [22] |
A.S. Panfilov, G.E. Grechnev, A.A. Lyogenkaya, et al., Phys. B 553 (2019) 80-87. DOI:10.1016/j.physb.2018.10.036 |
| [23] |
E.A. Kümmerle, G. Heger, et al., J. Solid State Chem. 147 (1999) 485-500. DOI:10.1006/jssc.1999.8403 |
| [24] |
T. Xie, X.D. Wang, M. Yao, et al., RSC Adv. 6 (2016) 20349-20356. DOI:10.1039/C5RA27890B |
| [25] |
B. Zhang, J. Liu, F. Shen, J. Phys. Chem. C 119 (2015) 15047-15055. DOI:10.1021/acs.jpcc.5b00645 |
| [26] |
H. Li, S. Liu, J. Yang, et al., Fuel 260 (2020) 116289. DOI:10.1016/j.fuel.2019.116289 |
| [27] |
M. Gupta, A. Kumar, A. Sagdeo, P.R. Sagdeo, J. Phys. Chem. C 125 (2021) 2648-2658. DOI:10.1021/acs.jpcc.0c09133 |
| [28] |
D.E.P. Vanpoucke, P. Bultinck, S. Cottenier, V. Van Speybroeck, I. Van Driessche, J. Mater. Chem. A 2 (2014) 13723-13737. DOI:10.1039/C4TA02449D |
| [29] |
C. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci. 509 (2020) 145392. DOI:10.1016/j.apsusc.2020.145392 |
| [30] |
G. Bersuker, D.C. Gilmer, D. Veksler, et al., J. Appl. Phys. 110 (2011) 124518. DOI:10.1063/1.3671565 |
| [31] |
Y. Xue, D. Tian, C. Zeng, Y. Fu, K. Li, AIP Adv. 9 (2019) 125341. DOI:10.1063/1.5124317 |
| [32] |
J. Li, D. Wang, R. Guan, et al., ACS Sustain. Chem. Eng. 8 (2020) 18258-18265. DOI:10.1021/acssuschemeng.0c06775 |
| [33] |
W. Song, K. Xie, J. Wang, et al., Phys. Chem. Chem. Phys. 23 (2021) 10418-10428. DOI:10.1039/d1cp00690h |
| [34] |
X. Fu, H. Yang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 1089-1094. DOI:10.1016/j.cclet.2020.08.031 |
| [35] |
C. He, R. Wang, H. Yang, S. Li, L. Fu, Appl. Surf. Sci. 507 (2020) 145076. DOI:10.1016/j.apsusc.2019.145076 |
| [36] |
W. Li, Q. Jiang, D. Li, Z. Ao, T. An, Chin. Chem. Lett. 32 (2021) 2803-2806. DOI:10.1016/j.cclet.2021.01.026 |
| [37] |
J.K. Nørskov, T. Bligaard, A. Logadottir, J. Electrochem. Soc. 152 (2005) J23. DOI:10.1149/1.1856988 |
| [38] |
H. Yang, C. He, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3203-3206. DOI:10.1016/j.cclet.2021.03.038 |
| [39] |
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J. 414 (2021) 128857. DOI:10.1016/j.cej.2021.128857 |
| [40] |
G. Liu, J. Zhou, W. Zhao, Z. Ao, T. An, Chin. Chem. Lett. 31 (2020) 1966-1969. DOI:10.1016/j.cclet.2019.12.023 |
| [41] |
A. Oaks, D. Yun, B. Ye, W.Y. Chen, J.F. Stubbins, J. Nucl. Mater. 414 (2011) 145-149. DOI:10.1016/j.jnucmat.2011.02.030 |
| [42] |
W. Song, L. Fu, C. He, et al., Adv. Theory Simul. (2021) 2100044. DOI:10.1002/adts.202100044 |
| [43] |
J. Zamudio-García, L.D. Santos-Gómez, J.M. Porras-Vázquez, E.R. Losilla, D. Marrero-López, J. Alloy. Compd. 816 (2020) 152600. DOI:10.1016/j.jallcom.2019.152600 |
| [44] |
M. Firdos, F. Hussain, M. Imran, et al., Mater. Res. Express 4 (2017) 106301. DOI:10.1088/2053-1591/aa896e |
| [45] |
D. Zhou, C. Li, F. Yin, et al., Chin. Chem. Lett. 31 (2020) 2325-2329. DOI:10.1016/j.cclet.2020.04.045 |
| [46] |
D. Tian, K. Li, Y. Wei, et al., Phys. Chem. Chem. Phys. 20 (2018) 11912-11929. DOI:10.1039/C7CP08376A |
| [47] |
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149-3154. DOI:10.1016/j.cclet.2021.02.046 |
| [48] |
L. Fu, L. Yan, L. Lin, et al., J. Alloy. Compd. 875 (2021) 159907. DOI:10.1016/j.jallcom.2021.159907 |
| [49] |
X. Li, J. Liu, J. Huang, et al., Acta Phys. Chim. Sin. 37 (2021) 2010030. |
| [50] |
W. Song, J. Wang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3137-3142. DOI:10.1016/j.cclet.2021.02.043 |
| [51] |
M.A. Légaré, G. Bélanger-Chabot, R.D. Dewhurst, et al., Science 359 (2018) 896-900. DOI:10.1126/science.aaq1684 |
| [52] |
C. Ling, X. Niu, Q. Li, A. Du, J. Wang, J. Am. Chem. Soc. 140 (2018) 14161-14168. DOI:10.1021/jacs.8b07472 |
| [53] |
J.C. Liu, X.L. Ma, Y. Li, et al., Nat. Commun. 9 (2018) 1610. DOI:10.1038/s41467-018-03795-8 |
| [54] |
Y. Guo, Y. Cheng, Q. Li, K. Chu, J. Energy Chem. 56 (2021) 259-263. DOI:10.1016/j.jechem.2020.07.055 |
| [55] |
D. Sun, H. Bai, Y. Zhao, et al., ACS Appl. Mater. Interfaces 12 (2020) 52763-52770. DOI:10.1021/acsami.0c16337 |
| [56] |
J. Zhao, X. Ren, X. Li, et al., Nanoscale 11 (2019) 4231-4235. DOI:10.1039/c8nr10401h |
| [57] |
Y.K. Peng, K.M. Lee, C.C. Ting, M.W. Hsu, C.Y. Liu, J. Mater. Chem. A. DOI:10.1039/C9TA09777E |