 2022, Vol. 33
2022, Vol. 33 


b Institute of Energy, Hefei Comprehensive National Science Center, Hefei 230031, China;
c School of Energy and Environment Science, Anhui University of Technology, Maanshan 243032, China;
d Department of Applied Physics, The Hong Kong Polytechnic University, Hong Kong, China
Hydrogen has long been regarded as clean energy carrier for sustainable but intermittent energy storage and value-added feedstock for modern chemical manufacture [1, 2]. Water electrolysis is an innovative approach towards hydrogen production without carbon emission, yet its industrial application still fails to be realized owing to the low electricity conversion efficiency [3-5]. Related to cathodic hydrogen evolution reaction (HER), anodic oxygen evolution reaction (OER) undergoes a four-electron transfer process with sluggish reaction kinetics, typically determining the overall efficiency [6, 7]. Current state-of-the-art OER electrocatalysts are noble-metal-based oxide such as RuO2 and IrO2, which still require an overpotential of > 300 mV to reach the benchmark current density of 10 mA/cm2 [8]. Moreover, their high-cost and low-durability nature largely hinder the exploration for substantial large-scale implementations. In this regard, it is of high urgency to seek the OER electrocatalyst alternatives with low cost, high activity and good durability [9, 10].
Given the tunable electronic states, 3d transition-metal-based materials have received increasing attention as OER electrocatalysts, especially in alkaline electrolyte [9-15]. Among the materials, transition metal phosphide is a promising class of candidates due to their approximately zero-valent metallic feature with high electronic conductivity [16-19]. Extensive approaches have been devoted to developing the OER electrocatalyst advances from the viewpoint of both crystal and electronic structures, including metal/phosphorus component regulation and exotic atomic doping. Regardless of these glorious accomplishments, it still confronts a great challenge to improve the intrinsic activity of metal phosphides. Fundamental studies unveil that the electronic states of catalytically active sites play the decisive roles in molecular adsorption and activation (i.e., chemical affinity of adsorbates) to govern the intrinsic activity [5-7]. Such a goal of electronic manipulation can be achieved by the construction of nanoscale interface through rational heterostructure design [20-22]. The key knob is to ensure the chemical interaction at the interface for efficient electron coupling and transfer.
Inspired by the above considerations, here we develop a co-phosphorization strategy for constructing a VPO4-Ni2P heterostructure anchored on nickel foam (NF) with strongly interfacial interaction. Phosphate is regarded as a critical species for OER activity improvement [17, 23]. In out heterostructure, VPO4 serves as a robust electronic modulator to withdraw electron from Ni2P via the strong electron coupling, giving a key contribution to interfacial electron transfer from Ni2P to VPO4. Such a heterostructure renders the electron density deficiency in Ni2P with more oxidized Ni species. Theoretical simulations reveal that the chemical affinities of oxygen intermediates in OER pathway (i.e., *OH, *O and *OOH) are rationalized to lower the overall energy barrier for activity enhancement. As a result, this advanced heterostructure achieves a remarkable OER activity with a low overpotential of 289 mV at a large current density of 350 mA/cm2 and a high turnover frequency (TOF) value of 0.378 s–1 at an overpotential of 290 mV in alkaline media.
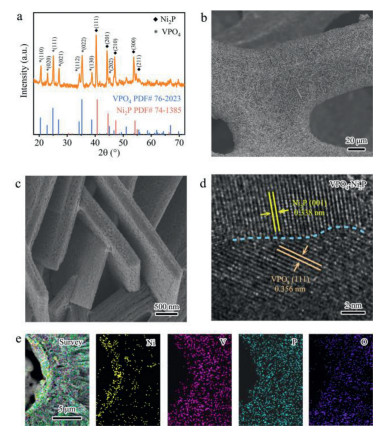
The VPO4-Ni2P heterostructure catalyst anchored on NF (Fig. S1 in Supporting information) is synthesized through a two-step procedure as illustrated in Scheme S1 (Supporting information). A vanadium oxide (VOx) precursor is firstly synthesized on NF via the hydrothermal method (step Ⅰ in Scheme S1). SEM images (Fig. S2 in Supporting information) manifest that VOx is grown vertically on NF substrate with the nanosheet structure. Then the as-obtained VOx/NF precursor undergoes a thermal treatment to achieve the co-phosphorization using NaH2PO2 as phosphorus source (Step Ⅱ in Scheme S1). Phase characterization through XRD (Fig. 1a) indicates that this phosphorization process not only converts VOx into VPO4 (PDF#76-2023) but also functionalizes the surface of NF with Ni2P (PDF#74-1385), thereby forming a VPO4-Ni2P heterostructure. Considering the tight generation of VOx precursor on NF, such a co-phosphorization strategy can offer a strongly chemical interaction at the interface. Similar phosphorization treatment of bare VOx and NF yields VPO4 and Ni2P/NF samples for references, respectively (Figs. S3 and S4 in Supporting information). SEM images of VPO4-Ni2P heterostructure (Figs. 1b and c) manifest that VPO4 maintains the well-defined nanosheet structure. To be different, the porous structure is observable for VPO4 (Fig. 1c) compared to pristine VOx with smooth surface. This unique porous feature can facilitate the diffusion of reactants on catalyst surface to promote the reactant diffusion and mass transfer efficiency [24, 25].

|
Download:
|
| Fig. 1. Structural characterizations of VPO4-Ni2P heterostructure. (a) XRD pattern. (b) Low-resolution and (c) high-resolution SEM images. (d) HRTEM image. (e) SEM-based EDS mapping profiles of Ni (yellow), V (violet), P (cyan) and O (blue) elements, respectively. | |
{kind=link}
To resolve the interface of VPO4 and Ni2P, HRTEM is further employed to characterize the boundary of this heterostructure. Fig. 1d clearly shows two lattice fringe spacings of 0.356 and 0.338 nm, which can be indexed to the (111) plane of VPO4 and (001) plane of Ni2P, respectively. The small lattice mismatch enables the tight lattice interaction between VPO4 and Ni2P, which benefits the interfacial charge transfer. We also conduct EDS mapping to resolve the SEM-based elemental dispersion of VPO4-Ni2P sample. As shown in Fig. 1e, the aggregated dispersion of Ni and V signals at the interface further corroborates the heterojunction feature.
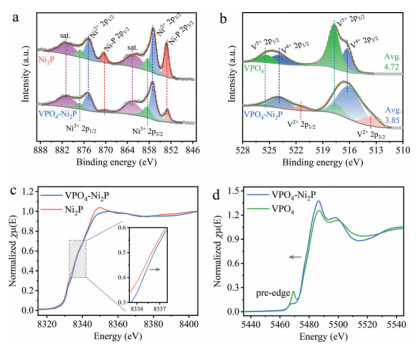
To resolve the chemical states of VPO4-Ni2P heterostructure, X-ray photoelectron spectroscopy (XPS) measurements are conducted. The deconvolution results of metal (Ni and V) 2p core-level spectrums show the distinct difference of valence states between VPO4-Ni2P heterostructure and pristine Ni2P or VPO4. In Ni 2p region (Fig. 2a), Niδ+ in phosphide (853.1 and 870.4 eV), oxidized Ni2+ (856.9 and 874.7 eV) and Ni3+ (858.5 and 877.1 eV) species are readily observable for both Ni2P and VPO4-Ni2P heterostructure, together with the shakeup satellites at 862.7 and 880.8 eV [17, 26]. Intuitively, the relative amount of Niδ+ species in VPO4-Ni2P heterostructure apparently decreases from 28% to 13% related to Ni2P, indicative of the existence of more oxidized Ni2+ and Ni3+ species (Fig. S5 in Supporting information). The increment of oxidized Ni species can supply more active sites for OER, which is supposed to give the vital contribution to the activity enhancement. On the other hand, contrary trend is recognized for V component. As indicated in Fig. 2b, the overall V 2p XPS spectrum of both 2p3/2 and 2p1/2 doublets can be deconvoluted to several peaks, which are assigned to V2+ (513.6 and 521 eV), V4+ (516.2 and 523.9 eV) and V5+ (517.7 and 525.5 eV) species, respectively [15, 27]. Compared with reference VPO4, the predominant vanadium species of VPO4-Ni2P heterostructure changes from V5+ to V4+ (Fig. S6 in Supporting information), together with an average valence reduction from +4.72 to +3.85. Such a valence variation for both Ni and V components manifests the strong chemical interaction at the interface of Ni2P and VPO4, giving the robust electron transfer from Ni2P to VPO4.

|
Download:
|
| Fig. 2. Valence state investigations of Ni2P-VPO4 heterostructure. (a) High-resolution Ni 2p and (b) V 2p XPS spectra. (c) Normalized Ni and (d) V K-edge XANES spectra. | |
{kind=link}
This electronic interaction observation is further consolidated by synchrotron radiation-based X-ray absorption near edge structure (XANES) spectra. As shown in normalized Ni K-edge XANES spectra (Fig. 2c), the absorption edge of VPO4-Ni2P heterostructure shifts towards the higher photon energy, suggesting the increased Ni valence. Concomitantly, V K-edge XANES spectra of VPO4-Ni2P heterostructure exhibits a red-shift absorption edge (Fig. 2d). To be specific, the pre-edge absorption predominantly originates from the distorted symmetry of octahedral V5+O6 moiety, whereas the significantly lower intensity in VPO4-Ni2P heterostructure related to VPO4 reference signifies the valence decrease (i.e., V4+ and/or V2+ species) [28]. As the electronic state of catalytic sites can alter the chemical affinity of adsorbed oxygen species, it is highly anticipated that the interfacial charge regulation in VPO4-Ni2P heterostructure can have the profound influence on OER performance.
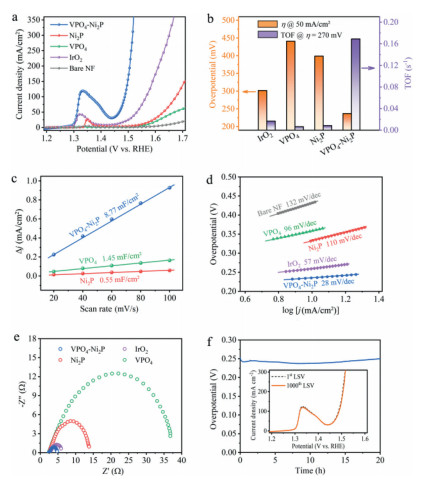
Upon acquiring the structural information, we are now in a position to assess the OER activity of our VPO4-Ni2P heterostructure. The electrochemical OER performance of VPO4-Ni2P heterostructure catalyst is evaluated in a standard three-electrode system using O2-saturated 1 mol/L KOH as electrolyte (see Experimental Section in Supporting information). Pristine Ni2P and VPO4 supported by NF are also tested for comparison, together with benchmark IrO2. Fig. 3a displays the collected linear sweep voltammogram (LSV) polarization curves of various catalysts, clearly exhibiting that VPO4-Ni2P heterostructure catalyst delivers the best OER activity. The overpotential (η) at the desired current density of 50 mA/cm2 is extracted to quantitatively elucidate the activity. To be specific, VPO4-Ni2P heterostructure requires an η of 237 mV (Fig. 3b, orange column), which is substantially lower than that of benchmark IrO2 (302 mV), Ni2P (399 mV), and VPO4 (441 mV) catalysts. Moreover, VPO4-Ni2P heterostructure reaches a high current density of 350 mA/cm2 at a low potential input of 1.519 V vs. reversible hydrogen electrode (RHE) (i.e., η = 289 mV). This remarkable performance with large current density indicates the promising potential from the viewpoint of practical application. It is noteworthy that the anodic redox peak prior to water oxidation exhibits distinct difference among VPO4-Ni2P heterostructure and pristine Ni2P catalysts. This redox can be assigned to the pre-oxidation of surface Ni species, which plays the critical roles in constructing active sites and determining the OER activity. The redox peak of VPO4-Ni2P heterostructure is much intense than that of Ni2P, showing more catalytically active sites for OER. Such a catalytic site increment can be interpreted by the robust charge transfer in interfacial Ni-(PO4)-V backbone, which turns more metallic Ni-P species to oxidized Ni species, as substantiated by XPS result (Fig. 2a).

|
Download:
|
| Fig. 3. OER activity evaluation. (a) LSV polarization curves at a scan rate of 5 mV/s. (b) Specific overpotential values at current density of 50 mA/cm2 (orange column) and TOF values at overpotential of 270 mV (violet column), respectively. (c) The determined Cdl values. (d) Tafel plots derived from polarization curves in Fig. 3a. (e) Nyquist plots at 1.5 V vs. RHE. (f) Chronopotentiometry curve of VPO4-Ni2P heterostructure catalyst at the current density of 10 mA/cm2 for stability test. Inset is LSV polarization curve after 1000 CV cycles. | |
{kind=link}
Considering the increased active site amount, we normalize the current density with veritably active sites to give the turnover frequency (TOF) values for assessing the intrinsic activity more fairly (Fig. S7 in Supporting information). As shown in Fig. 3b (violet column), VPO4-Ni2P heterostructure delivers a TOF value of 0.378 s–1 at η = 290 mV, 14.6, 26.7 and 45.3 times higher than benchmark IrO2, Ni2P and VPO4 catalysts, respectively. Such a high TOF value manifests that the intrinsic activity of catalytic sites is also significantly improved by the construction of heterojunction between VPO4 and Ni2P. Moreover, a negative shift of redox peak (~20 mV) is also observable for Ni2P-VPO4 heterostructure related to Ni2P reference. Owing to the alkaline condition, the catalyst surface is typically covered by the pre-adsorbed hydroxyl species, and the Ni species pre-oxidation is always accompanied by hydroxyl deprotonation. This negative shift implies the facilitated Ni pre-oxidation and pre-adsorbed hydroxyl deprotonation. We rationalize that this facilitated redox electrochemistry is also ascribed to interfacial electron transfer from Ni2P to VPO4, which renders electron density depletion in Ni2P (i.e., valence increment of Ni species). The altered Ni pre-oxidation feature is followed by a double-layer charging response to render the large electrochemical active surface area (ECSA). Specifically, the double-layer capacitance (Cdl) of Ni2P-VPO4 heterostructure is determined to be 8.77 mF/cm2 (Fig. 3c and Fig. S8 in Supporting information), 6.0- and 15.9-fold increase related to VPO4 and Ni2P, respectively.
To gain insights into reaction kinetics, the Tafel slopes are obtained as shown in Fig. 3d. The large Tafel slopes of 110 and 96 mV/dec for Ni2P and VPO4 catalysts indicate the sluggish OER kinetics. Remarkably, VPO4-Ni2P heterostructure endows an apparently reduced Tafel slope of 28 mV/dec, even lower than that of benchmark IrO2 (57 mV/dec), suggesting that OER is accelerated by interfacial electronic effect. Meanwhile, we also measure the electrochemical impedance spectroscopy (EIS) to probe the charge transfer efficiency at catalyst/electrolyte interface. The smallest semicircle of VPO4-Ni2P heterostructure in Nyquist plot (Fig. 3e) indicates the reduced charge transfer resistance (Rct) that accelerates electron transfer at catalyst/electrolyte interface. It is worth noting that our VPO4-Ni2P heterostructure catalyst exhibits the competitive OER activity in comparison with the recently reported phosphide-based OER electrocatalyst advances (Table S1 in Supporting information).
The catalytic stability is also a vitally important parameter to evaluate the priority of catalyst candidate. To this end, we perform both chronopotentiometry and CV measurements for VPO4-Ni2P heterostructure catalyst. A 20-h chronopotentiometry test shows that an η below 250 mV is consecutively maintained to achieve a desired current density of 10 mA/cm2 (Fig. 3f). Meanwhile, LSV polarization curve after 1000 CV cycles also demonstrates the outstanding potential-current response without perceptible activity decline compared with the initial one (inset in Fig. 3f). Both the measurements declare the high stability of our VPO4-Ni2P heterostructure catalyst towards alkaline water oxidation. Structural characterizations after the durability tests demonstrate that the VPO4-Ni2P catalyst well retains the phase and porous nanosheet morphology (see XRD pattern and SEM image in Fig. S9 in Supporting information). Furthermore, XPS measurements of surface metallic elements reveal that the Ni and V elements are oxidized to higher oxidation states as compared with the pristine ones (Fig. S10 in Supporting information). This valence variation coincides with the pre-oxidation typically observed in electrochemical experiments, suggesting the nature of surface metallic species as catalytic sites in water oxidation. In particular, the Ni-P species are oxidized to Nix+ species that play the role as catalytic sites.
To further look into the fundamental origin of OER activity enhancement towards VPO4-Ni2P heterostructure, density functional theory (DFT) calculations are performed to construct the theoretical model for this heterojunction, resolving the electronic states and simulating the OER pathway. Ni2P(200) and VPO4(022) surfaces (Fig. S11 in Supporting information) are chosen to build the heterostructure based on the experimental observation. A stable atomic structure of the established VPO4-Ni2P heterostructure is schemed in Fig. 4a. It is recognized that the O atoms in VPO4 bind strongly with the Ni or P atoms in Ni2P. Such a chemical interaction will give rise to the interfacial charge redistribution. Owing to the larger electronegativity of O element than Ni or P element, it can be predicated that VPO4 overlayer obtains electrons from Ni2P. This conjecture is corroborated by charge density analysis. The computed isosurfaces of charge density difference (Fig. 4b) clearly demonstrate the electron depletion at Ni2P surface (cyan color) and electron aggregation at VPO4 surface (olive color), respectively. Bader charge analysis further quantitatively consolidates that this charge redistribution results in electron shift of 5.579|e| from Ni2P to VPO4. The computed results well coincide with the experimental observations (i.e., XPS and XANES results in Fig. 2).

|
Download:
|
| Fig. 4. DFT simulations. (a) The atomic schematic illustration of stable VPO4-Ni2P heterostructure. (b) Isosurfaces of charge density difference. Differential charge density by first-principles simulations illustrates the increase (olive color) and decrease (cyan color) of electron distributions. (c) PDOS diagrams of Ni2P, VPO4 and VPO4-Ni2P heterostructure models. (d) Free energy diagram of OER pathway on Ni2P and VPO4-Ni2P heterostructure models. | |
{kind=link}
Furthermore, we examine the electronic structure of the heterostructure. According to the total density of state (TDOS) diagram (Fig. S12 in Supporting information), bare Ni2P shows the metallic nature as the electronic state crosses the Fermi level (EF), whilst an obvious energy band is observable around EF for bare VPO4. This electronic feature indicates that charge transfer is largely impeded in VPO4, leading to the low catalytic activity. After constructing VPO4-Ni2P heterostructure, the substantially increased electron density at EF manifests the improvement of electrical conductivity, further facilitating the electron transfer between catalyst surface and adsorbates. Specifically, their projected density of state (PDOS) diagrams are depicted in Fig. 4c. It is recognized that the electron density increment for VPO4-Ni2P heterostructure is mainly contributed by V 3d orbitals. Since VPO4 receives electrons from Ni2P owing to interfacial charge transfer, EF shifts toward V 3d orbitals and finally a proportion of unoccupied 3d orbitals of V is filled. Moreover, the Ni d-band center in VPO4-Ni2P heterostructure (–2.11 eV vs. EF) is computed to downshift related to that in bare Ni2P (–2.09 eV vs. EF). Such an electronic regulation is also believed to tailor the chemical affinities of oxygen intermediates at catalyst surface, playing a vital role in the catalytic activity [29-31].
To look into the impact on reaction process, we investigate the adsorption behaviors of key intermediates (i.e., *OH, *O and *OOH) on bare Ni2P and VPO4-Ni2P heterostructure surfaces, respectively (Table S2 in Supporting information). Fig. 4d shows the Gibbs free energy diagram of the OER 4e– pathway based on the well-established adsorbate evolution mechanism (i.e., H2O(l) → *OH → *O → *OOH → O2(g)) [32]. On Ni2P(200) surface, the rate-limiting step (RLS) of OER is step Ⅳ to form O2 molecule, since *OOH is adsorbed too strongly on the surface. The free energy change of RLS is determined to be 2.53 eV (i.e., overpotential of 1.30 V). Differently, as for VPO4-Ni2P heterostructure surface, the chemical affinities of oxygen intermediates are rationalized by the regulated electronic structure, weakening their binding strengths at catalytic site. As a result, the RLS of OER on VPO4-Ni2P heterostructure surface turns to oxygen coupling process (i.e., step Ⅲ). The related free energy change is reduced to 1.97 eV (i.e., overpotential of 0.74 V), manifesting the activity improvement for VPO4-Ni2P heterostructure catalyst.
In summary, we have constructed a VPO4-Ni2P heterostructure on nickel foam substrate as a high-performance OER electrocatalytic advance. Both experimental and theoretical studies uncover the strong coupling interaction at the interface of VPO4 and Ni2P. Specifically, VPO4 acts as robust electronic modulator to trigger the electron transfer from Ni2P to VPO4 through interfacial Ni-(PO4)-V backbone, giving rise to electron deficiency in Ni2P. Such an electronic modulation contributes more catalytic sites. More importantly, the chemical affinities of oxygen intermediates are rationalized to weaken the binding strength on VPO4-Ni2P heterostructure, altering the rate-limiting step to improve the intrinsic activity towards alkaline water oxidation. As a result, the VPO4-Ni2P heterostructure delivers a remarkable OER activity with a low overpotential of 289 mV at a large current density of 350 mA/cm2 and a high TOF value of 0.378 s–1 at an overpotential of 290 mV. This work offers new insights into seeking low-cost, high-performance OER electrocatalyst alternatives and also reaffirms the importance of interfacial engineering towards catalyst design at atomic precision.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported in part by National Key R & D Program of China (Nos. 2020YFA0406103, 2017YFA0207301), National Natural Science Foundation of China (Nos. 21725102, 91961106, U1832156, 22075267, 21803002), Science and Technological Fund of Anhui Province for Outstanding Youth (No. 2008085J05), Youth Innovation Promotion Association of CAS (No. 2019444), Young Elite Scientist Sponsorship Program by CAST, MOST (No. 2018YFA0208603), Users with Excellence Program of Hefei Science Center CAS (No. 2020HSC-UE003) and Postdoc Matching Fund Scheme of the Hong Kong Polytechnic University (No. 1-W144). XAFS measurements were performed at the beamline 1W1B of the BSRF and beamline BL14W1 of SSRF. XPS experiments were performed at the photoemission endstations (BL10B) in NSRL. We thank the support from USTC Center for Micro- and Nanoscale Research and Fabrication.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.05.011.
| [1] |
J.A. Turner, Science 305 (2004) 972-974. DOI:10.1126/science.1103197 |
| [2] |
A.L. Goff, V. Artero, B. Jousselme, et al., Science 326 (2009) 1384-1387. DOI:10.1126/science.1179773 |
| [3] |
J. Kibsgaard, I. Chorkendorff, Nat. Energy 4 (2019) 430-433. DOI:10.1038/s41560-019-0407-1 |
| [4] |
I. Roger, M.A. Shipman, M.D. Symes, Nat. Rev. Chem. 1 (2017) 0003. DOI:10.1038/s41570-016-0003 |
| [5] |
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) eaad4998. DOI:10.1126/science.aad4998 |
| [6] |
N.T. Suen, S.F. Hung, Q. Quan, et al., Chem. Soc. Rev. 46 (2017) 337-365. DOI:10.1039/C6CS00328A |
| [7] |
J. Suntivich, K.J. May, H.A. Gasteiger, J.B. Goodenough, Y. Shao-Horn, Science 334 (2011) 1383-1385. DOI:10.1126/science.1212858 |
| [8] |
Q. Shi, C. Zhu, D. Du, Y. Lin, Chem. Soc. Rev. 48 (2019) 3181-3192. DOI:10.1039/C8CS00671G |
| [9] |
B.M. Hunter, H.B. Gray, A.M. Müller, Chem. Rev. 116 (2016) 14120-14136. DOI:10.1021/acs.chemrev.6b00398 |
| [10] |
C. Hu, L. Zhang, J. Gong, Energ. Environ. Sci. 12 (2019) 2620-2645. DOI:10.1039/C9EE01202H |
| [11] |
B. Zhang, X. Zheng, O. Voznyy, et al., Science 352 (2016) 333-337. DOI:10.1126/science.aaf1525 |
| [12] |
Z.F. Huang, J. Song, Y. Du, et al., Nat. Energy 4 (2019) 329-338. DOI:10.1038/s41560-019-0355-9 |
| [13] |
N. Zhang, X. Feng, D. Rao, et al., Nat. Commun. 11 (2020) 4066. DOI:10.1038/s41467-020-17934-7 |
| [14] |
B. Zhang, L. Wang, Z. Cao, et al., Nat. Catal. 3 (2020) 985-992. DOI:10.1038/s41929-020-00525-6 |
| [15] |
J. Jiang, F. Sun, S. Zhou, et al., Nat. Commun. 9 (2018) 2885. DOI:10.1038/s41467-018-05341-y |
| [16] |
X. Zheng, B. Zhang, P. De Luna, et al., Nat. Chem. 10 (2018) 149-154. DOI:10.1038/nchem.2886 |
| [17] |
F. Hu, S. Zhu, S. Chen, et al., Adv. Mater. 29 (2017) 1606570. DOI:10.1002/adma.201606570 |
| [18] |
H. Huang, C. Yu, C. Zhao, et al., Nano Energy 34 (2017) 472-480. DOI:10.1016/j.nanoen.2017.03.016 |
| [19] |
T. Liu, A. Li, C. Wang, et al., Adv. Mater. 30 (2018) 1803590. DOI:10.1002/adma.201803590 |
| [20] |
Y. Bai, W. Zhang, Z. Zhang, et al., J. Am. Chem. Soc. 136 (2014) 14650-14653. DOI:10.1021/ja506269y |
| [21] |
B. Qiu, C. Wang, N. Zhang, et al., ACS Catal. 9 (2019) 6484-6490. DOI:10.1021/acscatal.9b01819 |
| [22] |
Z.W. Gao, J.Y. Liu, X.M. Chen, et al., Adv. Mater. 31 (2019) 1804769. DOI:10.1002/adma.201804769 |
| [23] |
H. Zhou, F. Yu, J. Sun, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 5607-5611. DOI:10.1073/pnas.1701562114 |
| [24] |
J. Qi, W. Zhang, R. Xiang, et al., Adv. Sci. 2 (2015) 1500199. DOI:10.1002/advs.201500199 |
| [25] |
X. Peng, L. Zhang, Z. Chen, et al., Adv. Mater. 31 (2019) 1900341. DOI:10.1002/adma.201900341 |
| [26] |
B. Qiu, L. Cai, Y. Wang, et al., Adv. Funct. Mater. 28 (2018) 1706008. DOI:10.1002/adfm.201706008 |
| [27] |
E. Hryha, E. Rutqvist, L. Nyborg, Surf. Interface Anal. 44 (2012) 1022-1025. DOI:10.1002/sia.3844 |
| [28] |
D.A. McKeown, I.S. Muller, K.S. Matlack, I.L. Pegg, J. Non-Cryst. Solids 298 (2002) 160-175. DOI:10.1016/S0022-3093(02)00945-6 |
| [29] |
J.K. Nørskov, F. Abild-Pedersen, F. Studt, T. Bligaard, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 937-943. DOI:10.1073/pnas.1006652108 |
| [30] |
H. Wang, J. Wang, Y. Pi, et al., Angew. Chem. Int. Ed. 58 (2019) 2316-2320. DOI:10.1002/anie.201812545 |
| [31] |
A. Grimaud, K.J. May, C.E. Carlton, et al., Nat. Commun. 4 (2013) 2439. DOI:10.1038/ncomms3439 |
| [32] |
I.C. Man, H.Y. Su, F. Calle-Vallejo, et al., ChemCatChem 3 (2011) 1159-1165. DOI:10.1002/cctc.201000397 |