 2022, Vol. 33
2022, Vol. 33 


b School of Civil Engineering, Tianjin University, Tianjin 300072, China;
c Beijing Key Laboratory of Aqueous Typical Pollutants Control and Water Quality Safeguard, Department of Municipal and Environmental Engineering, School of Civil Engineering, Beijing Jiaotong University, Beijing 100044, China;
d Institute of Surface-Earth System Science, School of Earth System Science, Tianjin University, Tianjin 300072, China;
e Tianjin Key Laboratory of Earth Critical Zone Science and Sustainable Development in Bohai Rim, Tianjin University, Tianjin 300072, China
A large amount of prescribed/fed pharmaceuticals are excreted and enter into surface aquatic system [1], resulting in huge threats to the ecosystem due to their toxicity and potential to induce drug resistance [2]. Pharmaceuticals have been detected in various water matrices such as drinking water, surface water, groundwater, and wastewater at ng/L to µg/L levels [3-6]. Meanwhile, most pharmaceuticals are not persistent and undergo transformation through different pathways, resulting in different abundance and environmental effects [7, 8]. Therefore, elucidation of their transformation kinetics and mechanism will benefit the management of pharmaceutical contamination and risk assessment.
Structural transformation induced by environmental reactive species is one of the dominant pathways of pharmaceutical transformation in both natural waters and wastewaters [9, 10]. Hydroxyl radical (•OH) has drawn the most attention due to its high oxidation potential (E0(•OH/H2O) = 1.9 − 2.7 V) and low selectivity [11-13]. Carbonate radical (CO3•−), which is normally generated from the reaction between •OH (or other reactive species such as triplet-excited state of dissolved organic matter (DOM) [14]) and HCO3−/CO32−, is now increasingly investigated due to its prevalent occurrence [9, 15, 16]. Carbonate radical is electrophilic and able to degrade pharmaceuticals (E0 (CO3•−/CO32−) = 1.63 V at pH 8.4), but is more chemical-structurally selective than •OH [17]. The background effect is considered lower than •OH and its steady-state concentration is usually higher, reaching above 10−14 mol/L level depending on the DOM content and pH condition in sunlit surface water and around 10−12 mol/L in advanced oxidation processes [18]. This compensates its lower oxidation power and makes it a significant contributor to pharmaceutical transformation. In addition, when treating some pharmaceutical-containing waste matrices such as source separated urine, CO3•− was reported as the dominant reactive species (at around 10−10 mol/L) over •OH and contributed the most to the degradation of some antibiotics [15].
Compared with •OH, fewer studies have reported the reactivity of CO3•− towards pharmaceuticals. To date, there are only 49 pharmaceuticals with reported rate constants [19]. In addition, although quantitative structure-activity relationships (QSARs) have been established to predict the reactivity of •OH [20], O3 [21] and sulfate radical (SO4•−) [22], the QSAR models are rather scarce on CO3•− reactivity. Lack of reactivity information hinders the assessment of CO3•− contribution to transformation pathways of pharmaceuticals in aquatic systems.
The establishment of QSARs is highly dependent on the selection of the relevant molecular descriptors. There are more than 5000 descriptors currently, each representing a portion of the molecular properties [23]. Selecting appropriate descriptors that can capture the entire picture of the reactivity is of huge difficulty. A simpler method without need of complicated descriptor is preferable. Molecular fingerprints (MF) encode structural features of molecules as binary vectors and has been adopted in developing QSAR models to predict ligand biological activity [24] and toxicity [25, 26]. Zhang et al. first used deep neural network (DNN) combined with MF to predict the reactivity of •OH towards organics [23]. It showed that the obtained MF-DNN models had comparable prediction accuracies to the traditional QSARs.
Therefore, in this study, DNN combined with MF was applied to establish a model for CO3•− reactivity prediction. A data processing method (MaxMin) was applied to improve the accuracy of the model [27]. The reactivity of human pharmaceuticals and veterinary drugs towards CO3•− was predicted.
A dataset containing 231 organic compounds and their second-order rate constants with CO3•− were extracted from the literatures [19, 28]. Because CO3•− reacts with organic molecules primarily through electron transfer mechanism, there is likely non-negligible influence of the speciation of compounds towards reacting with CO3•−. Therefore, pKa values were collected and the exact forms of the compounds under the pH conditions that the rate constants were determined. This led to a total of 252 distinct chemical forms applied for analysis (see Datasets excel file in Supporting information). Generally, for those compound/form with several reported rate constants, an average value was used.
Chemical diversity and the application domain analysis is essential to build a structure-based predictive model. The dataset of 231 organic compounds covered element C, H, O, N, S, F, Cl, Br and P. The MaxMin algorithm (MaxMinPicker function in RDkit toolbox (https://www.rdkit.org/)), based on fingerprint similarity calculations [27], constructed a group of sub dataset which obtained the highest structural diversity in the overall data set. The chemical space distribution (defined by number of molecular features in MF and rate constants in this study) of the training set and test set was further evaluated. As shown in Fig. S1 (Supporting information), they shared a similar chemical space. The application domain was evaluated using Tanimoto Similarity to avoid prediction for compounds differing significantly from the training set. Each compound was converted to MF, based on which the average distance Dave and the standard deviation of distance σ were calculated. Z is an arbitrary parameter to control the significance level, which was set at 0.5 in this study. Therefore, the application domain threshold, DT, was defined by the equation (DT = Dave + Zσ). For the training dataset, the DT value was 0.933. If the distance between a compound in the test or validation datasets and its nearest neighbor in the training set exceeded DT, the prediction was considered unreliable.
We converted SMILES (simplified molecular-input line-entry system) strings of the compounds/forms to Morgan MF by the RDKit toolbox. The generated MF were binary vectors, each represents the presence of a certain structural feature. The length of MF was adjustable. Longer MF length stores more structural features. The Mordred application in python package [29] was used to calculate 1832 MD including constitutional, topological, geometrical descriptors, etc.
We primarily evaluated the performance of the developed models by the root mean square error (RMSE), coefficient of determination (R2), accuracy of prediction (ACC). Description of these index is provided in Text S1 (Supporting information).
To achieve initial screening results of optimal set-up for rates prediction, we conducted an orthogonal experiment with an array of MF parameters and DNN structures. Radius and vector length are two essential MF parameters for Morgan fingerprint. As shown in Fig. S2 (Supporting information), varying radius from 1 to 3 and vector length from 512 to 2048 did not result in significant difference in the RMSE values. Interestingly, within a single cell (for a certain DNN structure), the lowest RMSE often appeared at the combination of the smallest radius (i.e., r = 1) and the smallest vector length (i.e., vl = 512). This may indicate the reaction rates between organic compounds with carbonate radical are likely determined by local chemical properties instead of those at molecular level. And putting more diverse features (vector length) in the model input may diluted the importance of structures contributing to the difference in rates.
In contrast to MF parameters, upgrading the DNN structure from simple to complex (i.e., increasing the numbers of hidden layers and neurons) considerably improved the model performance. The optimal hyperparameters were around three hidden layers with 1024 neurons fully connected. Further increase of the complexity of model structure did not yield lower RMSE.
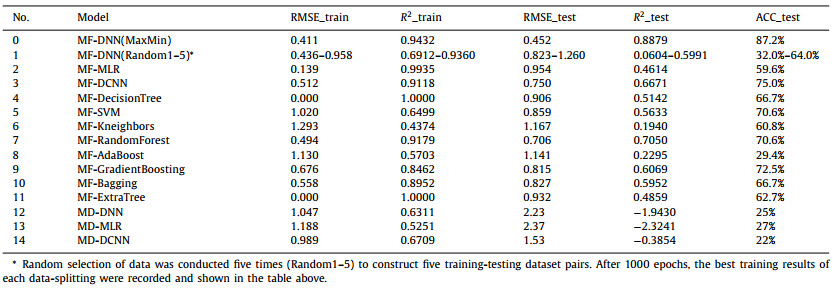
Therefore, we took this structure as base structure and further adjusted model hyperparameters. The model with the best fitting results for validation dataset is shown in Fig. 1. The RMSE value, R2 and accuracy were 0.452, 0.888 and 87.2%, respectively.

|
Download:
|
| Fig. 1. (A) The scatterplot of the predicted vs the experimental values of log kCO3•- for training, test and external validation datasets. (B) The structural of optimized DNN. | |
External validation was performed using the data from literatures [16, 30, 31] and NIST database, in which most of the compounds are of environmental relevance. As shown in Fig. 1, this model presented a satisfactory prediction for the external validation dataset with accuracy of 10/13. The ones out of EG 0.2/5 boundaries were still within one-order of magnitude.
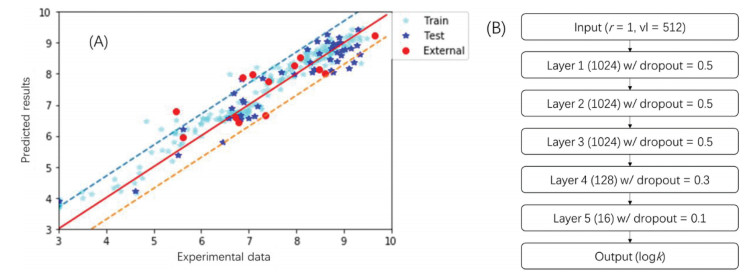
To evaluate if the combination of MaxMin, MF and DNN yielded the best results for carbonate radical rate prediction, we compared MaxMin with random selection for training set construction, MF with MD for model input, and DNN with multivariable linear regression, supporting vector machine, and other commonly used machine learning methods.
The application of MaxMin dissimilarity-based selection (MaxMin) is expected to pick the most structurally diverse subset of molecules in a given dataset [27]. However, it was not clear if MaxMin could result in better predictions for the rates of carbonate radical. Therefore, we tested the performance of MF-DNN using MaxMin selection and random selection, which is the most commonly applied in constructing training dataset. After 1000 epochs, the results of the best models were recorded (Table 1). Although the DT values of the training set of these two methods were approximately the same (DT = 0.933–0.939), the performance of MaxMin significantly excelled those of random selection. Indeed, the prediction accuracy for MaxMin was over 20% higher than those of random selection (Table 1).
|
|
Table 1 Performance of models for the training set and test set using MF, MD and different modeling methods. |
{kind=link}
Table 1 also shows the performance of models for the training set and test set using MD and different modeling methods. Comparing MF and MD (Model No. 0, 2, 3 with No. 12, 13, 14), the results of MF-based models were considerably superior to MD-based models. Indeed, the accuracy of the best performance of MD-based models was below 30%, significantly lower than that of MF-based models.
Multivariable linear regression (MLR), deep neural network (DNN), and deep convolutional neural network (DCNN), random forest, supporting vector machine (SVM), etc., are among the mostly commonly applied regression algorithms in machine learning. For MF-based models, 1024 bits were initially applied as input data structure linearly for DNN model, whereas a 32 × 32 matrix constructed from 1024 bits was used for DCNN model. The MD-DNN model took 1832 descriptors as input data, whereas MD was reconstructed into a 43 × 43 matrix with zero filling in empty space for DCNN models. The output layers of all models gave a single value which was used to compare with the target second-order rate constant (log kCO3•−). Comparing these 10 algorithms, DNN almost excelled in all quality index. Multivariable linear regression (MLR), decision tree and extra tree methods showed perfect fitting for training set, whereas these models performed poorly on test set, suggesting significant over-fitting results. DCNN yielded the second-best results of all modeling methods, whereas DCNN consumed significant high computing resource than DNN. For example, a 100-epoch running time on a laptop was over 30 min for DCNN, comparing with less than 30 s for DNN.
Carbonate radical is primarily produced in sunlit surface water and advanced water treatment processes. Therefore, we chose pharmaceuticals used in human medicine and veterinary practice as the target chemicals because of their high adverse impact on eco-system and high probability to be exposed to carbonate radical. A group of 1576 drugs for human medicine was obtained from the subset of FDA drugs for sale in ZINC15 database (http://zinc.docking.org/substances/subsets/fda+for-sale); and a group of 390 drugs with distinct molecular forms for veterinary use was extracted from the Green Book (USDA, https://www.fda.gov/animal-veterinary/products/approved-animal-drug-products-green-book). The total 1966 molecules had passed the check for model application domain (i.e., the distances between target molecule and train dataset were within DT), suggesting the prediction of rate constants of these pharmaceuticals was with high confidence.
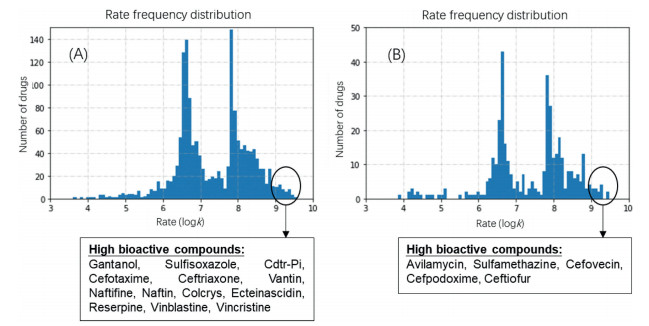
As shown in Fig. 2, the frequency distribution of rate constants grouped into two distinct peaks at around logk of 6.5 and 8.0. Forty-six drugs (39 in human medicine, 7 for veterinary medicine) were identified with rate constants higher than 109 L mol−1 s−1, suggesting that carbonate radical may significantly contribute to the overall environmental attenuation. It is worth noticing that 18 out of these carbonate-radical-reactive drugs are of high bioactive properties, such as anti-microbial effects and acute toxicity (Fig. 2). The chemical structures are provided in Figs. S3 and S4 (Supporting information). This finding may suggest future research on the environmental fate of those drugs should be conducted with the emphasize on reactions involving carbonate radical.

|
Download:
|
| Fig. 2. The predicted rate constants of pharmaceuticals for human medicine (A), and veterinary use (B). High bioactive compounds with rate constants higher than 109 L mol−1s−1 were also listed. | |
{kind=link}
In conclusion, this study combined deep neural network with molecular fingerprints (MF-DNN) to construct a QSAR model, which successfully predicted the second-order rate constants between carbonate radical and organics. A new data processing method (MaxMin), which was designed to select a sub-group of molecules with maximized structural diversity, was applied to construct training dataset. This method helps to overcome the limited numbers of experimental data for carbonate radical. Applying this MF-DNN model, reaction rates between carbonate radical and pharmaceuticals used in human medicine (1576) and veterinary practice (390) were estimated. Among them, 46 drugs were identified as fast-reacting compounds, suggesting the important relations of their environmental fate with carbonate radical.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 41703101) and the Beijing Outstanding Young Scientist Program (No. BJJWZYJH01201910004016).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.06.061.
| [1] |
K. Kümmerer, Annu. Rev. Environ. Resour. 35 (2010) 57-75. DOI:10.1146/annurev-environ-052809-161223 |
| [2] |
W. Calero-Caceres, A. Melgarejo, M. Colomer-Lluch, et al., Environ. Sci. Technol. 48 (2014) 7602. DOI:10.1021/es501851s |
| [3] |
T. Zhang, B. Li, Crit. Rev. Environ. Sci. Technol. 41 (2011) 951-998. DOI:10.1080/10643380903392692 |
| [4] |
M.J. Benotti, R.A. Trenholm, B.J. Vanderford, et al., Environ. Sci. Technol. 43 (2009) 597-603. DOI:10.1021/es801845a |
| [5] |
S.R. Hughes, P. Kay, L.E. Brown, Environ. Sci. Technol. 47 (2013) 661-677. DOI:10.1021/es3030148 |
| [6] |
Y. Luo, W. Guo, H.H. Ngo, et al., Sci. Total Environ. 473-474 (2014) 619-641. DOI:10.1016/j.scitotenv.2013.12.065 |
| [7] |
R. Zhang, Y. Yang, C.H. Huang, et al., Environ. Sci. Technol. 50 (2016) 2573. DOI:10.1021/acs.est.5b05604 |
| [8] |
C.M. de Jongh, P.J.F. Kooij, P. De Voogt, et al., Sci. Total Environ. 427-428 (2012) 70-77. DOI:10.1016/j.scitotenv.2012.04.010 |
| [9] |
D. Vione, M. Minella, V. Maurino, et al., Chem. (Easton) 20 (2015) 10590-10606. DOI:10.1002/chem.201400413 |
| [10] |
R. Zhang, Y. Yang, C.H. Huang, et al., Water Res. 103 (2016) 283-292. DOI:10.1016/j.watres.2016.07.037 |
| [11] |
H. Yao, P. Sun, D. Minakata, et al., Environ. Sci. Technol. 47 (2013) 4581-4589. DOI:10.1021/es3052685 |
| [12] |
B.A. Wols, C.H.M. Hofman-Caris, D.J.H. Harmsen, et al., Water Res. 47 (2013) 5876-5888. DOI:10.1016/j.watres.2013.07.008 |
| [13] |
G.V. Buxton, C.L. Greenstock, W.P. Helman, et al., Phys. Chem. Ref. Data 17 (1988) 513-886. DOI:10.1063/1.555805 |
| [14] |
J. Wang, K. Wang, L. Zhang, et al., Water Res. 197 (2021) 117078. DOI:10.1016/j.watres.2021.117078 |
| [15] |
R. Zhang, P. Sun, T.H. Boyer, et al., Environ. Sci. Technol. 49 (2015) 3056. DOI:10.1021/es504799n |
| [16] |
Y. Zhou, C. Chen, K. Guo, et al., Water Res. 185 (2020) 116231. DOI:10.1016/j.watres.2020.116231 |
| [17] |
Z. Zuo, Z. Cai, Y. Katsumura, et al., Radiat. Phys. Chem. 55 (1999) 15-23. DOI:10.1016/S0969-806X(98)00308-9 |
| [18] |
S. Yan, Y. Liu, L. Lian, et al., Water Res. 161 (2019) 288-296. DOI:10.1016/j.watres.2019.06.002 |
| [19] |
L. Wojnárovits, T. Tóth, E. Takács, Sci. Total Environ. 717 (2020) 137219. DOI:10.1016/j.scitotenv.2020.137219 |
| [20] |
T.N.G. Borhani, M. Saniedanesh, M. Bagheri, et al., Water Res. 98 (2016) 344-353. DOI:10.1016/j.watres.2016.04.038 |
| [21] |
Y. Huang, T. Li, S. Zheng, et al., Sci. Total Environ. 715 (2020) 136816. DOI:10.1016/j.scitotenv.2020.136816 |
| [22] |
R. Xiao, T. Ye, Z. Wei, et al., Environ. Sci. Technol. 49 (2015) 13394-13402. DOI:10.1021/acs.est.5b03078 |
| [23] |
S. Zhong, J. Hu, X. Fan, et al., J. Hazard. Mater. 383 (2020) 121141. DOI:10.1016/j.jhazmat.2019.121141 |
| [24] |
K.Z. Myint, L. Wang, Q. Tong, et al., Mol. Pharm. 9 (2012) 2912-2923. DOI:10.1021/mp300237z |
| [25] |
K. Mansouri, A. Abdelaziz, A. Rybacka, et al., Environ. Health Perspect. 124 (2016) 1023-1033. DOI:10.1289/ehp.1510267 |
| [26] |
Y. Wu, G. Wang, Int. J. Mol. Sci. 19 (2018) 2358. DOI:10.3390/ijms19082358 |
| [27] |
M. Ashton, J. Barnard, F. Casset, et al., Mol. Inform. 21 (2003) 598-604. |
| [28] |
P. Neta, R.E. Huie, A.B. Ross, J. Phys. Chem. Ref. Data 17 (1988) 1027-1284. DOI:10.1063/1.555808 |
| [29] |
H. Moriwaki, Y.S. Tian, N. Kawashita, et al., J. Cheminform. 10 (2018) 4. DOI:10.1186/s13321-018-0258-y |
| [30] |
P. Sun, T. Meng, Z. Wang, et al., Environ. Sci. Technol. 53 (2019) 9024-9033. DOI:10.1021/acs.est.9b00749 |
| [31] |
Z. Hao, J. Ma, C. Miao, et al., Environ. Sci. Technol. 54 (2020) 10118-10127. DOI:10.1021/acs.est.0c03404 |