 2022, Vol. 33
2022, Vol. 33 


Nowadays, the energy crisis is one of the urgent issues faced by us. Sunlight-driven water splitting in artificial photosystem provides a promising strategy for sustainable and clean hydrogen fuel production [1-5]. Water oxidation is a half and crucial step in water splitting, consisting of releasing four protons and four electrons and dissociating the molecular dioxygen. During the last few decades, extraordinary efforts have been devoted to this challenging reaction [6-12]. Ru-based water oxidation catalysts (WOCs) are most studied among homogeneous water oxidation communities [13-25]. However, considering the cost, low toxicity and availability, first-row transition metal-based complexes are believed to be better candidates for WOCs. Even though significant progress has been made in this field [24-32], the ultimate goal of application in artificial photosystem still needs more efficient, stable and robust WOCs. To achieve this goal, a thorough mechanistic understanding by combing experiments and computation is essential. Quantum chemical calculations are playing more and more critical roles in this area [33-35].
The commercially available vitamin B12 has been reported to be an environmentally friendly catalyst for chemical reactions, such as dehalogenation, CO2 electrochemical reduction, hydrogenation of double bonds [36-39]. Because of the nontoxic and rich redox chemical property, vitamin B12 has attracted more and more attention in the catalytic community. Recently, Verpoort and co-workers reported that vitamin B12 could function as a homogeneous WOC electrochemically in pH 7.0 phosphate buffer (NaPi) solution with an overpotential of 0.58 V [40]. UV-vis, ESI-MS and FTIR experiments suggested that the catalyst remains its molecular structure up to 11 h [40]. The onset potential was observed at 1.2 V vs. Ag/AgCl, together with a more significant current density of water oxidation at around 1.5 V vs. Ag/AgCl. The linear relationship between the catalytic current and the catalyst concentration was also established, indicating a unimolecular mechanism. Furthermore, the increase of the concentration of NaPi leads to the linear growth of the current density in the working conditions, which suggested that phosphate ions may play an essential role in water oxidation [40].
In the present work, DFT calculations were used to investigate the mechanism of water oxidation catalyzed by vitamin B12 and address the role of phosphate ion in this chemical process. The methods used here are to a large extent similar to those in our previous water oxidation studies [33, 41, 42], and the details are shown in Supporting information. To construct the whole energy diagram, a reference potential of 1.72 V vs. SHE (corresponding to 1.5 V vs. Ag/AgCl) was used.
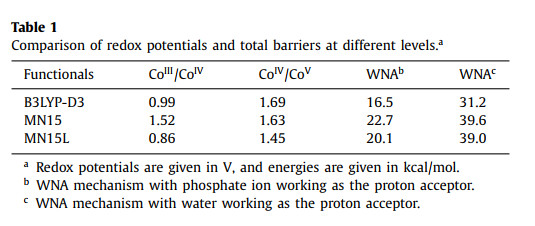
We first investigated the penta-coordinated complex 1 (CoIII-OH2, Fig. 1). The corrin ring has four nitrogen atoms coordinating with the cobalt center in the equatorial positions. An imidazole nitrogen atom is interacting with the metal center in the axial direction. The axial water molecule does not coordinate with the metal, instead interacts with the complex via two hydrogen bonds with the amide group of the ligand (Fig. 1). These two hydrogen bond distances are 2.03 and 1.90 Å, respectively. 1 prefers to be a triplet, while the singlet lies at +5.5 kcal/mol relative to the triplet. As shown in Fig. 1 and Fig. S2 (Supporting information), the spin density in 1 is partially delocalized on the ligand (total spin density of 0.91). Even though the formal oxidation state of the metal is CoIII, the electronic structure of 1 (L•-CoII) would be better described as a low-spin CoII center (SCo = 1/2) interacting with a corrin ligand radical cation (SL = 1/2). The coordination of water to cobalt to form a hexa-coordinated complex 1' (Fig. S3 in Supporting information) is 3.6 kcal/mol higher in energy.

|
Download:
|
| Fig. 1. Optimized structures of 1. Distances are shown in Å in red. Mulliken spin densities on selected atoms are shown in black. The multiplicity is shown in superscript. Hydrogen atoms of methyl are not shown for clarity. | |
The oxidation of 1 to form 2 (formally CoIV-OH, Fig. S4 in Supporting information) is coupled with the release of one proton from the water ligand, since the pKa of 2pt (the protonated form of 2, Fig. S2) was calculated to be -5.9. 2 is a doublet with the quartet state lying at +16.3 kcal/mol. The redox potential was calculated to be 0.99 V. Taking 1.72 V as a reference, this step is exergonic by 16.8 kcal/mol (Fig. 2). The spin delocalization on the ligand (spin density of 1.25) in 2 indicates that its electronic structure (L•-CoIII-OH) can be described as a low-spin CoIII (SCo = 0) with a ligand radical cation (SL = 1/2) (Fig. S2). The Co-O1 bond is 1.88 Å in 2.

|
Download:
|
| Fig. 2. Gibbs energy profile for water oxidation at the B3LYP-D3 level. Optimized TS1 (top right), TS1' and TS1'' (bottom left) in broken singlet are also shown. The imaginary frequencies of TS1, TS1' and TS1'' were 1237.2 i cm−1, 1164.0 i cm−1 and 610.7 i cm−1, respectively. Some of the unimportant atoms are not shown for clarity. Distances are shown in Å in red. Mulliken spin densities on selected atoms are shown in black. | |
Next, a proton-coupled electron transfer (PCET) process of 2 leads to the generation of 3 (formally CoV=O, Fig. S3) with a redox potential of 1.69 V. The calculated value is fully consistent with the experimental observation of 1.72 V [40]. The pKa of 3pt (the protonated form of 3) was calculated to be 1.8 (Fig. S2, 3pt). The Mulliken spin population analysis (1.0 on the corrin and 1.0 on O1) suggested that the electronic structure of 3pt (L•-CoIII-O•) can be interpreted as a low-spin CoIII (SCo = 0) interacting with an oxyl radical (SO = 1/2) as well as a ligand radical (SL = 1/2) ferromagnetically, which is reminiscent of previously reported, cobalt hangman-corroles WOC [43, 44]. 3 is a triplet, while the broken-symmetry singlet is slightly higher in energy, locating at +0.8 kcal/mol above. The Co-O1 distance in 3 is 1.81 Å which is somewhat shorter than that in 2.
The formally CoV=O species has been widely proposed to trigger the O-O bond formation for cobalt-based WOCs [34, 44-47]. Here, two possible mechanistic scenarios for the O-O bond formation were explored. First, a water molecule performs the nucleophilic attack on the oxyl moiety of 3 to form the O-O bond, during which one proton is transferred from the attacking water molecule to phosphate ion, either HPO42− or H2PO4− in working conditions (vide infra). The use of phosphate as a base has been suggested by the experiment and other metal-based water oxidation processes [34, 46-50]. Second, water molecules in the aqueous solution could also function as a proton acceptor.
The transition state (TS1) for the water nucleophilic attack (WNA) pathway with the HPO42− behaving as a proton acceptor is shown in Fig. 2. TS1 prefers to be a broken-symmetry singlet, and a spin crossing from the triplet to the broken-symmetry singlet is required to prompt the O-O bond formation. In TS1, the atomic spin density on the oxyl radical is 0.49α, while the ligand has the opposite spin density of 0.53β, which provides the most efficient electronic state for the O-O bond formation. (Fig. 2) During the O-O bond formation, the attacking water molecule provides a pair of electrons to form the O-O bond [33, 51]. The α-electron from the nucleophilic water molecule transfers to the ligand, while the proton is transferred to the phosphate. This can be considered an uncommon PCET process in which proton and electron are delivered to different acceptors. The β-electron left could easily combine with the oxyl radial to afford the O-O bond, which is similar to our previous work on the iron WOC [41]. TS1 was confirmed to have only one imaginary frequency of 1237.2 i cm−1, which corresponds to the O-O bond formation and proton transfer from water to phosphate. At TS1, the nascent O1-O2 bond distance is 1.96 Å. TS1 has a barrier of +16.5 kcal/mol above the 3. The triplet state lies at +9.5 kcal/mol relative to the broken-symmetry singlet. Downhill from TS1, the singlet intermediate Int1 with a hydroperoxide coordinated to CoIII (Fig. S4) is generated. In Int1, the O1-O2 bond length is 1.45 Å, and the Co-O1 distance increases from 1.81 Å in 3 to 1.83 Å in Int1. Int1 lies at +4.6 kcal/mol relative to 3. When H2PO4− was used as the proton acceptor, the barrier was calculated to be 17.9 kcal/mol (TS1', Fig. 2), which is 1.4 kcal/mol higher than the HPO42− assisted pathway. The NBO calculations were carried out for the transition states, and the NBO charge for HPO42− moiety in TS1 is −1.75, while in TS1' the charge is calculated to be −0.84 for H2PO4− moiety (Table S2 in Supporting information). This explains the favorability for the former pathway for WNA. In TS1', the O1-O2 distance is 0.03 Å shorter than that in TS1. In the working condition (pH 7.0, 0.1 mol/L NaPi), the concertation of H2PO4− and HPO42− are 0.062 mol/L and 0.038 mol/L, respectively. In any case, the HPO42− assisted pathway is preferred.
Alternatively, the transition state (TS1'') with water molecules featuring as the proton acceptor is shown in Fig. 2. In this configuration, three more water molecules were added to solvate the generated hydronium explicitly [41, 52, 53]. TS1'' was confirmed as a real transition state with only one imaginary frequency of 610.7 i cm−1. TS1'' prefers to be a broken-symmetry singlet, in which the oxyl radical and the corrin ligand have opposite spin populations (0.44 on the oxyl, while -0.44 on the ligand). The crucial O1-O2 distance is 1.89 Å at TS1''. The Co- O1 length increases to 1.86 Å from 1.81 Å in 3. The O2-H1 and O4-H1 distances are 1.02 Å and 1.58 Å, respectively. The barrier for this pathway was calculated to be 31.2 kcal/mol, which is much higher than the phosphate pathway (Fig. 2). The triplet TS1'' was calculated to be 8.5 kcal/mol higher than the broken singlet. This WNA process also generated the hydro-peroxide intermediate Int1, which joins the previous pathway.
From Int1, a PECT process leads to the formation of Int2 (Fig. S6 in Supporting information) with a redox potential of only 0.13 V. This demonstrated that this oxidation process is quite facile, with an exergonicity of 36.7 kcal/mol (Fig. 2). Int2 is a quartet state, while the doublet state is 3.0 kcal/mol higher. In Int2, the triplet dioxygen is already generated since the spin densities on O1 and O2 are 0.95 and 0.99, respectively. Meanwhile, the O2 dissociates from the cobalt center with the calculated Co-O1 distance of 3.26 Å. Furthermore, during the oxidation process, one electron is transferred from the hydro-peroxide moiety to the Co center with the formation of a low spin CoII, whose spin density is 0.99 (Fig. S4). The release of O2 from Int2 is followed by the binding of a water molecule to form the doublet intermediate Int3 (Fig. S4), with the quartet 12.1 kcal/mol higher in energy. This ligand exchange process is exergonic by 4.0 kcal/mol (Fig. 2). Finally, the last one-electron oxidation process from Int3 regenerates 1, with a calculated redox potential of 0.32 V. It should be mentioned that the ligand is involved in this oxidation process, as the spin is partially delocalized on the ligand in 1 (L•-CoII).
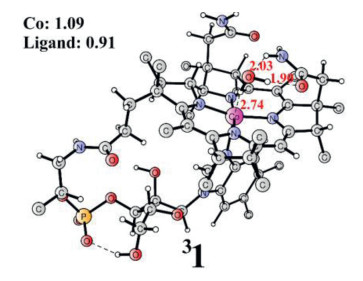
From Fig. 2, it can be seen that the O-O bond formation is the rate-determining step with a calculated total barrier of 16.5 kcal/mol at the B3LYP-D3 level. It should be mentioned that the use of different applied potentials may change the total barrier. When using a reference potential lower than 1.69 V (oxidation potential of 2 to 3), the total barrier will increase somewhat as the formation of 3 becomes endergonic. For example, when a reference potential of 1.4 V was used to set up the energy diagram, the total barrier increases to 23.2 kcal/mol (Table S1 in Supporting information). If the reference potential is higher than 1.69 V, the total barrier does not change as the formation of 3 is exergonic. 3 is the resting state for the O-O bond formation and its electronic structure is L•-CoIII-O•. Therefore, the catalytic activity of water oxidation is assisted by the redox non-innocent corrin ligand, which avoids the access of the high-valent metal center. The use of non-innocent ligands has been disclosed in other metal-based WOCs, a beneficial strategy in the catalytic water oxidation process [11, 41, 48, 54]. Furthermore, single-point calculations were performed using the MN15 and MN15L functionals [55, 56]. The calculated redox potentials and total barriers are summarized in Table 1. For the first oxidation process, the MN15 gives a relatively higher redox potential value than B3LYP-D3 and MN15L. Importantly, all functionals give similar results for the O-O bond formation. When HPO42− was used to accept the proton, the barrier is 14.7 kcal/mol lower than water receiving the proton at the B3LYP-D3 level. The barrier differences at the MN15 and MN15L levels are 16.9 kcal/mol and 18.9 kcal/mol, respectively. This demonstrated that HPO42− is a better base for the O-O bond formation in the water oxidation process.
|
|
Table 1 Comparison of redox potentials and total barriers at different levels.a |
{kind=link}
{kind=link}
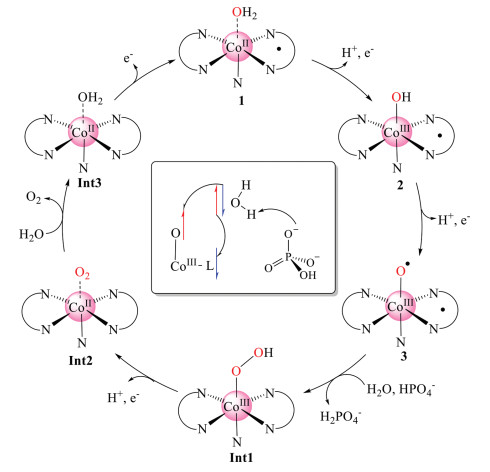
To conclude, we provide insights into the mechanism of water oxidation catalyzed by commercially available cobalamin (vitamin B12) through density functional calculations. The suggested catalytic cycle is summarized in Scheme 1, and the corresponding energy diagram is shown in Fig. 2. The starting point of the catalytic cycle is the formal CoIII-OH2 complex 1. Two sequential PCET processes result in the formation of a formal CoV=O intermediate 3, which can be described as L•-CoIII-O•. The redox non-innocent character of the corrin ligand in the water oxidation was illuminated. 3 initiates the O-O bond formation via WNA with the production of a hydro-peroxide intermediate. Importantly, HPO42− in the phosphate buffer was suggested to be the proton acceptor during the O-O bond formation. This step was suggested to be rate-determining with a total barrier of 16.5 kcal/mol. The alternative pathway with water accepting the proton during the O-O bond formation was calculated to be kinetically unfavorable since the barrier is 14.7 kcal/mol higher. The triplet dioxygen molecule dissociated from the cobalt center spontaneously after one PCET process from the CoIII hydro-peroxide intermediate. Finally, the second water molecule enters after the release of O2, followed by another one-electron oxidation process to regenerate 1.

|
Download:
|
| Scheme 1. Suggested water oxidation mechanism. Inset: The electron flow during the O-O bond formation is described. | |
{kind=link}
Declaration of competing interest
All authors declare that no conflict of interest exists.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21873031) and the National Key R & D Program of China (No. 2018YFA0903500).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.06.028.
| [1] |
B. Zhang, L. Sun, Chem. Soc. Rev. 48 (2019) 2216-2264. DOI:10.1039/c8cs00897c |
| [2] |
N. Wang, H. Zheng, W. Zhang, R. Cao, Chin. J. Catal. 39 (2018) 228-244. DOI:10.1016/S1872-2067(17)63001-8 |
| [3] |
M.D. Karkas, B. Akermark, Dalton Trans. 45 (2016) 14421-14461. DOI:10.1039/C6DT00809G |
| [4] |
M.D. Karkas, O. Verho, E.V. Johnston, B. Akermark, Chem. Rev. 114 (2014) 11863-12001. DOI:10.1021/cr400572f |
| [5] |
Y. Yang, Y. Tang, H. Jiang, et al., Chin. Chem. Lett. 30 (2019) 2089-2109. DOI:10.1016/j.cclet.2019.10.041 |
| [6] |
Q. Han, Y. Ding, Dalton Trans. 47 (2018) 8180-8188. DOI:10.1039/C8DT01291A |
| [7] |
P. Garrido-Barros, C. Gimbert-Surinach, R. Matheu, X. Sala, A. Llobet, Chem. Soc. Rev. 46 (2017) 6088-6098. DOI:10.1039/C7CS00248C |
| [8] |
J.D. Blakemore, R.H. Crabtree, G.W. Brudvig, Chem. Rev. 115 (2015) 12974-13005. DOI:10.1021/acs.chemrev.5b00122 |
| [9] |
B.M. Hunter, H.B. Gray, A.M. Muller, Chem. Rev. 116 (2016) 14120-14136. DOI:10.1021/acs.chemrev.6b00398 |
| [10] |
H. Wang, S. Zhu, J. Deng, et al., Chin. Chem. Lett. 32 (2021) 291-298. DOI:10.1016/j.cclet.2020.02.018 |
| [11] |
G. Wang, Z. Li, W. Wu, et al., Phys. Chem. Chem. Phys. 22 (2020) 24446-24454. DOI:10.1039/d0cp03925j |
| [12] |
G. Wang, Y. Zhi, L. Xia, et al., Phys. Status Solidi A 217 (2020) 1900859. DOI:10.1002/pssa.201900859 |
| [13] |
N. Vereshchuk, R. Matheu, J. Benet-Buchholz, et al., J. Am. Chem. Soc. 142 (2020) 5068-5077. DOI:10.1021/jacs.9b11935 |
| [14] |
S. Zhan, J.A. De Gracia Trivino, M.S.G. Ahlquist, J. Am. Chem. Soc. 141 (2019) 10247-10252. DOI:10.1021/jacs.9b02585 |
| [15] |
Q. Daniel, L. Duan, B.J.J. Timmer, et al., ACS Catal. 8 (2018) 4375-4382. DOI:10.1021/acscatal.7b03768 |
| [16] |
R. Matheu, M.Z. Ertem, J. Benet-Buchholz, et al., J. Am. Chem. Soc. 137 (2015) 10786-10795. DOI:10.1021/jacs.5b06541 |
| [17] |
S. Neudeck, S. Maji, I. Lopez, et al., J. Am. Chem. Soc. 136 (2014) 24-27. DOI:10.1021/ja409974b |
| [18] |
L. Duan, F. Bozoglian, S. Mandal, et al., Nat. Chem. 4 (2012) 418-423. DOI:10.1038/nchem.1301 |
| [19] |
D.J. Wasylenko, C. Ganesamoorthy, M.A. Henderson, et al., J. Am. Chem. Soc. 132 (2010) 16094-16106. DOI:10.1021/ja106108y |
| [20] |
S.W. Kohl, L. Weiner, L. Schwartsburd, et al., Science 324 (2009) 74-44. DOI:10.1126/science.1168600 |
| [21] |
L. Duan, A. Fischer, Y. Xu, L. Sun, J. Am. Chem. Soc. 131 (2009) 10397-10399. DOI:10.1021/ja9034686 |
| [22] |
J.J. Concepcion, J.W. Jurss, J.L. Templeton, T.J. Meyer, J. Am. Chem. Soc. 130 (2008) 16462-16463. DOI:10.1021/ja8059649 |
| [23] |
R. Zong, R.P. Thummel, J. Am. Chem. Soc. 127 (2005) 12802-12803. DOI:10.1021/ja054791m |
| [24] |
C. Sens, I. Romero, M. Rodriguez, et al., J. Am. Chem. Soc. 126 (2004) 7798-7899. DOI:10.1021/ja0486824 |
| [25] |
J.A. Gilbert, D.S. Eggleston, W.R. Murphy, et al., J. Am. Chem. Soc. 107 (1985) 3684-3670. |
| [26] |
Y. Li, R. Yao, Y. Chen, et al., Catalysts 10 (2020) 1-20. |
| [27] |
G. Maayan, N. Gluz, G. Christou, Nat. Catal. 1 (2018) 48-54. DOI:10.1038/s41929-017-0004-2 |
| [28] |
M. Okamura, M. Kondo, R. Kuga, et al., Nature 530 (2016) 465-468. DOI:10.1038/nature16529 |
| [29] |
P. Garrido-Barros, D. Moonshiram, M. Gil-Sepulcre, et al., J. Am. Chem. Soc. 142 (2020) 17434-17446. DOI:10.1021/jacs.0c06515 |
| [30] |
H.Y. Du, S.C. Chen, X.J. Su, L. Jiao, M.T. Zhang, J. Am. Chem. Soc. 140 (2018) 1557-1565. DOI:10.1021/jacs.8b00032 |
| [31] |
H. Lee, X. Wu, L. Sun, ChemSusChem 13 (2020) 3277-3282. DOI:10.1002/cssc.202000153 |
| [32] |
L. Wang, L. Duan, R.B. Ambre, et al., J. Catal. 335 (2016) 72-78. DOI:10.1016/j.jcat.2015.12.003 |
| [33] |
R.Z. Liao, P.E.M. Siegbahn, ChemSusChem 10 (2017) 4236-4263. DOI:10.1002/cssc.201701374 |
| [34] |
M. Schilling, S. Luber, Front. Chem. 6 (2018) 1-21. DOI:10.3389/fchem.2018.00001 |
| [35] |
A.V. Marenich, J. Ho, M.L. Coote, C.J. Cramer, D.G. Truhlar, Phys. Chem. Chem. Phys. 16 (2014) 15068-15106. DOI:10.1039/C4CP01572J |
| [36] |
M. Giedyk, K. Goliszewska, D. Gryko, Chem. Soc. Rev. 44 (2015) 3391-3404. DOI:10.1039/C5CS00165J |
| [37] |
K.L. Brown, Chem. Rev. 105 (2005) 2075-2149. DOI:10.1021/cr030720z |
| [38] |
R. Banerjee, S.W. Ragsdale, Annu. Rev. Biochem. 72 (2003) 209-247. DOI:10.1146/annurev.biochem.72.121801.161828 |
| [39] |
J. Chen, C. Karin, V.K. Priyank, et al., ACS Appl. Mater. Interfaces 12 (2020) 41288-41293. DOI:10.1021/acsami.0c10125 |
| [40] |
H.M. Shahadat, H.A. Younus, N. Ahmad, et al., Chem. Commun. 56 (2020) 1968-1971. DOI:10.1039/c9cc08838e |
| [41] |
Y.Y. Li, L.P. Tong, R.Z. Liao, Inorg. Chem. 57 (2018) 4590-4601. DOI:10.1021/acs.inorgchem.8b00333 |
| [42] |
R.Z. Liao, S. Masaoka, P.E.M. Siegbahn, ACS Catal. 8 (2018) 11671-11678. DOI:10.1021/acscatal.8b02791 |
| [43] |
D.K. Dogutan, J.R. McGuire, D.G. Nocera, J. Am. Chem. Soc. 133 (2011) 9178-9180. DOI:10.1021/ja202138m |
| [44] |
M.Z. Ertem, C.J. Cramer, Dalton Trans. 41 (2012) 12213-12219. DOI:10.1039/c2dt31871g |
| [45] |
A. Fernando, C.M. Aikens, J. Phys. Chem. C 119 (2015) 11072-11085. DOI:10.1021/jp511805x |
| [46] |
W. Lai, R. Cao, G. Dong, et al., J. Phys. Chem. Lett. 3 (2012) 2315-2319. DOI:10.1021/jz3008535 |
| [47] |
H. Lei, A. Han, F. Li, et al., Phys. Chem. Chem. Phys. 16 (2014) 1883-1893. DOI:10.1039/C3CP54361G |
| [48] |
J. Shi, Y.H. Guo, F. Xie, Q.F. Chen, M.T. Zhang, Angew. Chem. Int. Ed. 59 (2020) 4000-4008. DOI:10.1002/anie.201910614 |
| [49] |
M.K. Coggins, M.T. Zhang, Z. Chen, N. Song, T.J. Meyer, Angew. Chem. Int. Ed. 53 (2014) 12226-12230. DOI:10.1002/anie.201407131 |
| [50] |
H. Pan, L. Duan, R.Z. Liao, ChemCatChem 12 (2019) 219-226. |
| [51] |
D.W. Shaffer, Y. Xie, J.J. Concepcion, Chem. Soc. Rev. 46 (2017) 6170-6193. DOI:10.1039/C7CS00542C |
| [52] |
R.Z. Liao, X.C. Li, P.E.M. Siegbahn, Eur. J. Inorg. Chem. 4 (2014) 728-741. DOI:10.1002/ejic.201300710 |
| [53] |
M.Z. Ertem, L. Gagliardi, C.J. Cramer, Chem. Sci. 3 (2012) 1293-129. DOI:10.1039/c2sc01030e |
| [54] |
T. Wada, K. Tsuge, K. Tanaka, Inorg. Chem. 40 (2001) 329-337. DOI:10.1021/ic000552i |
| [55] |
H.S. Yu, X. He, D.G. Truhlar, J. Chem. Theory Comput. 12 (2016) 1280-1293. DOI:10.1021/acs.jctc.5b01082 |
| [56] |
H.S. Yu, X. He, S.L. Li, D.G. Truhlar, Chem. Sci. 7 (2016) 5032-5051. DOI:10.1039/C6SC00705H |