 2022, Vol. 33
2022, Vol. 33 


b Institute of Reproductive Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
With the development of genomics, increasing clinical evidence is showing that cancer and genetic diseases are closely related to DNA point mutations [1, 2]. Therefore, the detection of point mutations in driver genes is of great significance for the early diagnosis, treatment, and prognosis evaluation of cancer [3-5]. However, the abundance of human mutant DNA is generally low [6, 7]. Researchers have developed many methods for detecting low-abundance point mutations. The principle of the traditional detection method is that there is a thermodynamic difference between wild and mutant strands caused by a single mutant base, and then polymerase chain reaction (PCR) technology is used to distinguish between the two types of strands [8], such as droplet digital PCR [9], allele-specific blocker PCR [10, 11] and co-amplification at lower denaturation temperature-PCR (COLD-PCR) [12, 13]. However, the length of PCR amplicons is usually in the range of 100–200 bp, and the thermodynamic difference caused by a single mutant base is small; therefore, these methods are not highly sensitive. Moreover, these traditional detection methods cannot distinguish between target amplicons and non-specific products [14]. In recent years, researchers have developed next-generation sequencing (NGS)-based methods, such as Sanger sequencing, pyrosequencing, and single-molecule real-time (SMRT) sequencing, which have greatly reduced the sequencing detection limit to approximately 0.02% [15-18]. However, the equipment required by these methods is very expensive, thus increasing the cost of detection.
In view of the limitations of traditional detection and NGS methods, an increasing number of researchers are turning to post-PCR detection methods, such as DNA probes [19], molecular beacons [20], and toehold exchange probes [21]. Among them, DNA probes are widely used because of their strong specificity and high sensitivity. The working principle of the preliminary DNA probe is based on thermodynamic changes caused by a mismatch between the DNA probe and wild-type DNA [22]. Moreover, the practicality of enzymes provides additional tools for improving the identification ability of DNA probe-based post-PCR detection methods. By coupling the thermodynamic discrimination of the DNA probe with enzymatic signal amplification technology, the detection limit is greatly improved, and the method is simple, fast and cheap. Combination methods based on flap endonuclease [23], endonuclease IV [24] and lambda exonuclease [25] have been established, and the detection limit of such methods can reach 0.01%. The enzyme-linked DNA probe method requires the design of specific probes for each mutation site. This necessitates the synthesis of multiple probes, thereby increasing the cost of detection. Moreover, the constant adjustment of the probe sequence during the optimization process greatly increases the difficulty and cost of optimization.
Most cancers are caused by multiple mutations [26]. Therefore, the establishment of a multiplexed mutation detection method is of great significance for the diagnosis of cancer. Although there have been several methods for multiplexed mutation detection, such as a double-stranded DNA catalyzed strand displacement (dsCSD) system [27], High Resolution Melting (HRM) [28], and NGS method [15-18], they all have some limitations: the dsCSD system is low sensitivity and time-consuming; HRM is not robust; NGS is complicated and time-consuming. Due to these shortcomings, the above method has been greatly restricted in the application of multiple mutation detection. Therefore, there is an urgent need for the development of a universal, rapid, and low detection limit method for application to clinical genetic testing.
In this study, we developed a thermodynamics-guided interlocking two-way DNA cascade system for universal multiplexed mutation detection (TTI-CS) based on thermodynamic offset. This principle is illustrated in Fig. 1. The detection system has five DNA strands, probes labeled with fluorophores (FAM) and quenchers (BHQ), namely trigger, bridge, target, and blocker strands. Regions a and c of a trigger strand combine with a bridge strand to form a stable trigger/bridge complex. However, zone b in the middle of the trigger strand does not match the bridge strand, so a special bubble structure is formed in the trigger/bridge complex. Region b and parts of regions a and c of the trigger strand are also matched with a probe to form a stable trigger/probe complex. The stability of the trigger/bridge complex is greater than that of the trigger/probe complex. This leads to numerous trigger/bridge complexes and a very small amount of trigger/probe complexes at the initial stage of the system reaction process (room temperature, 37 ℃). However, when a target strand is added to the system, it binds bidirectionally to a trigger/bridge complex through the toehold zone (region d), to form a stable target/bridge/target complex. Moreover, the dissociated trigger strand combines with a free probe in the system, and numerous trigger/probe complexes are also formed. The enzyme then recognizes and binds to the trigger/probe complex to digest the probe. The two labeled fragments dissociate from the trigger strand, tearing apart the fluorophore-quencher (FAM-BHQ1) pair and releasing the fluorescent signal. In general, the reaction process of the system is divided into four reactions (reactions 1–4):
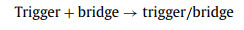
|
(1) |

|
(2) |

|
(3) |

|
(4) |

|
Download:
|
| Fig. 1. Schematic illustration of the working principle of TTI-CS. | |
{kind=link}
Reaction 4 is an enzymatic cyclic reaction. The trigger freed from a trigger/probe complex continues to combine with a probe strand in the system until all the free probes in the system are cleaved. Therefore, the system can generate a strong fluorescent signal. Reactions 2 and 3 are thermodynamically balanced and stable to achieve linkage, which is key to the working principle of the system. If the influence of the bubble structure is not considered, theoretically, the free energy change (∆G) is greater than zero for reactions 2 and 3. Therefore, reactions 2 and 3 process does not occur under normal conditions. For linkage to occur in these reactions, ∆G must be less than zero, which cannot be achieved by adjusting the number of trigger/bridge base pairing. With these facts in mind, we designed a bubble structure on the trigger/bridge complex, which greatly reduces the thermodynamic stability of the trigger/bridge complex. Thus, this bubble structure also greatly reduces the ∆G value of the entire reaction, enabling linkage in reactions 2 and 3. When target strands bind to a trigger/bridge complex, the formed trigger/target/bridge/target complex is extremely unstable, and the trigger dissociates. Owing to the existence of single-base mutations in mutant and wild strands, there is a significant difference in the thermodynamics. Thus, we designed the bridge strand to completely match the mutant-type (MT) and mismatch the wild-type (WT), so that the ∆G values of reactions 2 and 3 were much smaller for the former than for the latter strand. For the MT, it is easier to combine the trigger/bridge complex to achieve linkage in reactions 2 and 3; hence, it can be distinguished from the WT. Notably, to increase the distinguishing effect of the system on the two types of strands, we added a blocker strand that exactly matched the WT. Thus, by adjusting the length of the blocker sequence, we designed an MT process (reactions 2 and 3) with a ∆G that was significantly less than zero, and a WT process where ∆G was slightly less or greater than zero. For the MT, the linkage reaction process was as follows (reactions 5 and 6):

|
(5) |
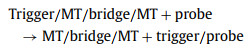
|
(6) |
For the WT, the above-mentioned linkage reaction process is difficult to occur. Therefore, when designing the trigger, bridge, and blocker sequences, we needed to determine whether the designed sequence was appropriate, according to the predicted ∆G values of the MT and WT reaction processes. In general, the system flexibly adjusts the thermodynamic balance of the reaction through the bubble structure, and a clear distinction between the MT and WT is achieved. It is also important to note that this system uses a strand migration reaction comprising two-way invasion of the target strand, which greatly accelerates the reaction speed and reduces detection time. In addition to adjusting the ∆G value of the reaction, the bubble structure also retains part of the sequence, creating a sequence that is unrelated to the target strand. Fig. 1 illustrates that the bubble sequence on the trigger strand and the bases on both sides of the bubble is completely unrelated to the target sequence. Therefore, when the sequence of the target strand changes, only the trigger and bridge sequences need to be changed accordingly. This can be achieved at a low cost and short process time.
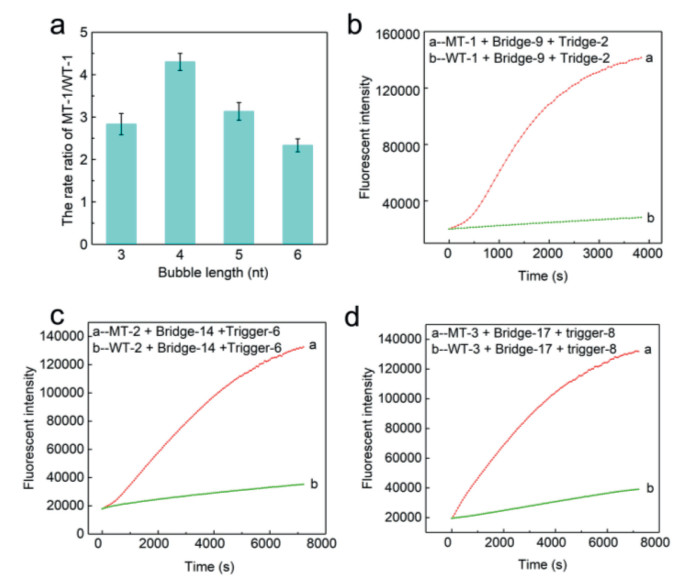
We proceeded to verify the feasibility of the designed detection system. Thus, taking the PTEN R130Q (G > A) mutation as the modeling target, we designed and synthesized mutant strand (MT-1); wild strand (WT-1); double-labeled (5′FAM and 3′BHQ1) fluorescent probes containing a purine/pyrimidine (AP) position in the middle of the strand (AP probe); and a series of a bridge, trigger, and blocker sequences (Table S1 in Supporting information). First, based on the ratio rates of MT-1 and WT-1 reaction determined by different trigger/bridge combinations, we chose the bridge-2/trigger-2 combination with the best effect, indicating that the sequence length of the bubble structure is 4 nt (Fig. 2a). We then further optimized the reaction conditions, including bridge, blocker sequences and concentration (Fig. S2 in the Supporting information). Subsequently, we used this method under the optimized conditions to measure the fluorescence curves of PTEN R130Q. As shown in Fig. 2b, the ratio of the rate of MT-1 and WT-1 was approximately 45. As mentioned earlier, TTI-CS adopts a two-way intrusion strand migration rapid reaction process, so that MT-1 and WT-1 can be distinguished within 60 min. Another highlight is that it is applicable to other enzyme-linked signal amplification strategies. Thus, we next used lambda exonuclease instead of endonuclease IV, while maintaining the other conditions constant (Fig. S3 in Supporting information). The gel electrophoresis experiment proved the occurrence of the above reaction process (Fig. S4 in Supporting information). Subsequently, we built a theoretical calculation model, to predict the best bridge, trigger, and blocker sequences by calculating ∆G during the entire reaction process of WT and MT, respectively (Supporting information).

|
Download:
|
| Fig. 2. (a) Optimization of the bubble length of bridge/trigger complex. (b-d) With the help of endonuclease IV, the fluorescence curves of PTEN R130Q, BRCA1/c.2082C > T, and EGFR L858R mutation points. | |
{kind=link}
As mentioned above, the most prominent advantage of this system is that for all the DNA point mutations, the probe can remain unchanged, which greatly facilitates multiplexed mutation detection and reduces the cost of detection. We also verified the unique versatility of the system through numerous experimental data. First, we chose BRCA1/c.2082C > T, EGFR L858R as modeling mutation sites and synthesized the corresponding MT, WT, bridge, trigger and blocker strands. Next, through theoretical calculation model and optimization of reaction conditions, we selected the best response sequence (Fig. S5 in Supporting information). Finally, under optimized conditions, we used endonuclease IV to measure the fluorescence curves of BRCA1/c.2082C > T and EGFR L858R. The ratio of the rise rates of the fluorescence signals of MT-2 and WT-2 for BRCA1/c.2082C > T was 9.2 (Fig. 2c), while that of MT-3 and WT-3 for EGFR L858R was approximately 8.5 (Fig. 2d). We also used lambda exonuclease to measure the fluorescence curves of BRCA1/c.2082C > T and EGFR L858R. The ratio of the rise rates of BRCA1/c.2082C > T was approximately 6.5, while that of EGFRL858R was 7 (Figs. S6a and b in Supporting information). The above experimental data fully prove that in multiple detections, compared to other detection methods, TTI-CS has the advantages of simplicity, speed, and efficiency.
Overall, we demonstrated the universality and discrimination ability of TTI-CS. We next evaluated the performance of this method in the detection of low-abundance mutations. Thus, taking PTEN R130Q, BRCA1/c.2082C > T, EGFR L858R, PTEN rs1473918395 and PTEN rs943020113 as the target mutations, we used the synthetic WT of each mutation point to dilute the MT, and prepared a series of mixed samples with a mutation abundance in the range of 100%–0.1%. We then used this system to measure the fluorescence curves of all the mixed samples at three mutation points. With the aid of endonuclease IV (Figs. 3a–c and Figs. S7a-b in Supporting information), the detection limit of these three mutation points all reached 0.1%, and all of them were completed in less than 1.5 h. With the assistance of lambda exonuclease (Figs. 3d–f), the detection limit of these three mutation points also reached 0.1%. This shows that TTI-CS method displays good low-abundance mutation detection performance, fast and stable. Moreover, compared with time-consuming NGS and dsCSD system methods, the detection time of TTI-CS is greatly shortened to 1.5 h in Fig. 3, and the most important thing is that the universality is well reflected by 5 mutation sites detected (Fig. 3 and Fig. S7 in Supporting information). Notably, TTI-CS based on bidirectional replacement requires the combination of two target strands on a bridge to form a trigger/target/bridge/target complex structure. Therefore, the detection limit of the TTI-CS is affected to a certain extent. In addition, in the detection of low-abundance mutations, the probability of two target strands binding to one bridge is greatly reduced, thereby affecting the detection limit of this method. However, this problem can be eliminated by increasing the concentration of the test target strand by various methods including PCR amplification technology, target strand enrichment technology, to improve the TTI-CS detection limit.

|
Download:
|
| Fig. 3. (a-c) With the help of endonuclease IV, the fluorescence curves for low abundance detection of PTEN R130Q, BRCA1/c.2082C > T, and EGFR L858R mutation points. (d-f) With the help of lambda exonuclease, the fluorescence curves for low abundance detection of PTEN R130Q, BRCA1/c.2082C > T, and EGFR L858R. | |
{kind=link}
Notably, it was necessary to explore the feasibility of the TTI-CS in clinical samples. The workflow is illustrated in Fig. 4a: First, we chose PTEN R130Q as the targeting mutation and removed the frozen cancer and normal endometrial tissue from liquid nitrogen. We then designed and synthesized the primers (FP and RP). Next, we extracted genomic DNA from the above-mentioned tissue and amplified the target DNA (WT-1-L and MT-1-L) by PCR. We then detected the PCR products by Sanger sequencing. The results revealed (Fig. S8 in Supporting information) that the mutation abundance of PTEN R130Q in a normal sample was 0%, while in a cancer sample was 50%. We proceeded by diluting the mutant DNA with genomic wild-type DNA to prepare a series of mixed DNA samples with a mutation abundance in the range of 50%–0.1%. We then used two-step (conventional and asymmetric) PCR and further processed the PCR product to obtain the target single strand. Finally, we detected the target single strand using TTI-CS. With the assistance of endonuclease IV, the TTI-CS detection limit was 0.1%, and was completed within 1.5 h (Fig. 4b), while with the aid of lambda exonuclease, the TTI-CS detection limit was 0.1% (Fig. 4c). In addition, to verify the accuracy of our test results, we used Sanger sequencing to detect the test target single strands (Fig. S9 in Supporting information). The detection results of TTI-CS are clearly consistent with the sanger sequencing results.

|
Download:
|
| Fig. 4. (a) Workflow of clinical sample detection. (b) With the help of endonuclease IV, the fluorescence curve for low abundance detection of PTEN R130Q. (c) With the help of lambda exonuclease, the fluorescence curve for low abundance detection of PTEN R130Q. | |
{kind=link}
In summary, we changed the thermodynamic balance and characteristics of the containment sequence by employing a bubble structure and used the principle of two-way invasion strand migration reaction to increase the reaction rate and constructed TTI-CS for low-abundance mutation detection. Using TTI-CS, we performed five-plex detection on three DNA point mutations of PTEN R130Q, BRCA1/c.2082C > T, EGFR L858R, PTEN rs1473918395 and PTEN rs943020113. The probe remained unchanged during the entire detection process and only the bridge and trigger sequences were replaced. This demonstrates the universality and low cost of the designed TTI-CS method. Our system displayed a detection limit of 0.1% in synthetic samples. TTI-CS was also successfully applied to the detection of endometrial cancer tissue samples, and the obtained PTEN R130Q detection limit was 0.1%. This proves the clinical applicability of the method. Overall, TTI-CS has the advantages of good versatility, high sensitivity, good stability and rapidity, and low cost.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Science and Technology Innovation Project of Hubei Province (No. 2019ACA138), the National Natural Science Foundation of China (Nos. 81871732 and 81974409).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.06.067.
| [1] |
Y.S. Chun, G. Passot, S. Yamashita, et al., Ann. Surg. 269 (2019) 917-923. DOI:10.1097/sla.0000000000002450 |
| [2] |
W. Zhang, Z. Ming, N. Chen, et al., Nanoscale 12 (2020) 20449-20455. DOI:10.1039/d0nr04691d |
| [3] |
M.T. Chang, S. Asthana, S.P. Gao, et al., Nat. Biotechnol. 34 (2016) 155-163. DOI:10.1038/nbt.3391 |
| [4] |
A. Letai, Nat. Med. 23 (2016) 1028-1035. |
| [5] |
X. Li, M. Ye, W. Zhang, et al., Biosens. Bioelectron. 126 (2016) 596-607. DOI:10.1177/0003122416641371 |
| [6] |
S.P. Shah, R.D. Morin, J. Khattra, et al., Nature 61 (2009) 809-813. DOI:10.1038/nature08489 |
| [7] |
K. Han, T.Y. Lee, D.E. Nikitopoulos, S.A. Soper, M.C. Murphy, Anal. Biochem. 417 (2011) 211-219. DOI:10.1016/j.ab.2011.06.030 |
| [8] |
S. Dall'Ozzo, C. Andres, P. Bardos, H. Watier, G. Thibault, J. Immunol. Methods 277 (2003) 185-192. DOI:10.1016/S0022-1759(03)00123-6 |
| [9] |
B.J. Hindson, K.D. Ness, D.A. Masquelier, et al., Anal. Chem. 83 (2011) 8604-8610. DOI:10.1021/ac202028g |
| [10] |
H. Chen, J. Zhang, H.Y. Chen, B. Su, D. Lu, Ann. Transl. Med. 8 (2020) 1509. DOI:10.21037/atm-20-6754 |
| [11] |
Q. Wang, D.P. Kontoyiannis, R. Li, et al., J. Clin. Microbiol. 57 (2019) e00604-e00619. |
| [12] |
S. Galbiati, A. Monguzzi, F. Damin, et al., J. Med. Genet. 53 (2016) 481-487. DOI:10.1136/jmedgenet-2015-103229 |
| [13] |
J. Li, L. Wang, H. Mamon, et al., Nat. Med. 14 (2008) 579-584. DOI:10.1038/nm1708 |
| [14] |
S.W. Tan, A. Ideris, A.R. Omar, K. Yusoff, M. Hair-Bejo, J. Virol. Methods 160 (2009) 149-156. DOI:10.1016/j.jviromet.2009.05.006 |
| [15] |
X. Xiao, A. Xu, J. Zhai, M. Zhao, Methods 64 (2013) 255-259. DOI:10.1016/j.ymeth.2013.06.031 |
| [16] |
J.M. Heather, B. Chain, Genomics 107 (2016) 1-8. DOI:10.1016/j.ygeno.2015.11.003 |
| [17] |
S. Ardui, A. Ameur, J.R. Vermeesch, M.S. Hestand, Nucleic Acids Res. 46 (2018) 2159-2168. DOI:10.1093/nar/gky066 |
| [18] |
M.A. Ihle, J. Fassunke, K. Konig, et al., BMC Cancer 14 (2014) 13. DOI:10.1186/1471-2407-14-13 |
| [19] |
D.X. Wang, J. Wang, Y.X. Cui, et al., Anal. Chem. 91 (2019) 13165-13173. DOI:10.1021/acs.analchem.9b03453 |
| [20] |
Q. Wang, L. Chen, Y. Long, H. Tian, J. Wu, Theranostics 3 (2013) 395-408. DOI:10.7150/thno.5935 |
| [21] |
H. Xu, W. Deng, F. Huang, et al., Chem. Commun. 50 (2014) 14171-14174. DOI:10.1039/C4CC07272C |
| [22] |
X. Chen, N. Liu, L. Liu, et al., Nat. Commun. 10 (2019) 4675. DOI:10.1038/s41467-019-12593-9 |
| [23] |
C.Y. Lee, H. Jang, H. Kim, et al., Mikrochim. Acta 186 (2019) 330. DOI:10.1109/icecs46596.2019.8964682 |
| [24] |
X. Tang, N. Chen, R. Liu, et al., Anal. Chim. Acta 134 (2020) 28-33. |
| [25] |
X. Weng, X. Xu, P. Huang, et al., Anal. Sci. 36 (2020) 697-702. DOI:10.2116/analsci.19p422 |
| [26] |
M.T. Chang, S. Asthana, S.P. Gao, et al., Nat. Biotechnol. 34 (2016) 155-163. DOI:10.1038/nbt.3391 |
| [27] |
N. Liu, X. Zhang, X. Tang, et al., Chem. Commun. 56 (2020) 14397-14400. DOI:10.1039/d0cc06216b |
| [28] |
H.L. Hondow, S.B. Fox, G. Mitchell, et al., BMC Cancer 265 (2011) 1471-2407. |