 2022, Vol. 33
2022, Vol. 33 


b CAS Key Laboratory of Energy Regulation Materials, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
Since trifluoromethyl group (CF3) has unique chemical stability and metabolic stability, introducing trifluoromethyl group into organic compounds at the specific position has been used as an important strategy for the modification of medicine, pesticides and functional molecular materials [1]. In the past two decades, much effort has been made to develop various methodologies for the preparation of CF3-containing compounds [2]. Among them, radical trifluoromethylation represents one of the most attractive strategies [3]. Trifluoromethylation reagents, such as TMSCF3 [4], CF3X (X = Br, I) [5], Langlois' reagent [6], Umemoto's reagents [7], Togni's reagents [8] are usually used as trifluoromethyl source. Traditional methods for generating CF3 radical required transition metals catalyst, chemical oxidant, or reductant, nevertheless, the use of these reagents inevitably generates environmentally hazardous waste.
In recent years, electrochemical organic synthesis represented a green and powerful method, has attracted much attention due to the avoiding of additional oxidation and reduction reagents in the reaction [9]. So far, an increasing number of examples about electrochemical trifluoromethylation reactions have been reported, such as C-H trifluoromethylation [10], difunctionalization of olefins [11], tandem trifluoromethylation [12]. However, most of these reactions have been realized by using CF3SO2Na and (CF3SO2)2Zn as trifluoromethylation reagent, which was oxidized to generate CF3 radical on the anode. Therefore, there is urgent need to study new reagents applied in electrochemical synthesis to accelerate the area.
Sulfonylhydrazides, which is easy to prepare, no bad smell, have been widely used in the reactions as arylation/alylating and thioetherification reagents [13]. In this regard, very recently, TfNHNHBoc has successfully been used as a trifluoromethylating for vicinal difunctionalization of terminal alkenes using TBHP or K2S2O8 as the oxidant and trifluoromethylthiolation of indoles [14]. Inspired by the previous CF3SO2Na-based electrochemical trifluoromethylation, herein, we investigated the potential of TfNHNHBoc as CF3 radical source under electrochemical conditions.
Trifluoromethylated indole compounds are the dominant heterocyclic scaffolds that are present in a variety of biologically active natural products and drugs [15]. Therefore, researchers are working to develop methods to provide various functionalized trifluoromethylated indole compounds [16]. In this context, the electrochemical trifluoromethylation/cyclization of N-arylacrylamide has significant advantages in the synthesis of fluoroalkylated indole compounds. Using CF3SO2Na (Scheme 1a), Zeng [17] developed an indirect electrochemical trifluoromethylation/cyclization of N-arylacrylamide using bromide as catalyst with a low loading. Subsequently, Mo [18] described Mn-mediated electrochemical trifluoromethylation/C(sp2)−H functionalization cascade for the synthesis of nitrogen heterocyclic compounds. Meanwhile, Ackermann [19] also successfully established catalyst-free, direct electrochemical tri- and difluoroalkylation/cyclization to prepare functionalized oxindoles and quinolinones. In view of this, we reported the direct electrochemical trifluoromethylation/cyclization using TfNHNHBoc as CF3 source for the first time (Scheme 1b).

|
Download:
|
| Scheme 1. Electrochemical trifluoromethylation/cyclization of N-arylacrylamide. | |
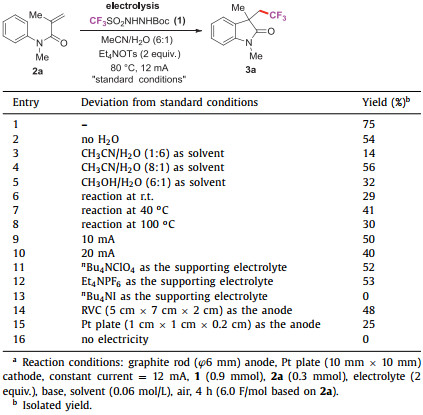
Initially, TfNHNHBoc (1) was prepared in one step by reacting commercially available Tf2O and NH2NHBoc [20]. To optimize the reaction conditions, N-arylacrylamide (2a) and TfNHNHBoc (1) were chosen as the model substrates. This reaction was carried out in an undivided cell equipped with a graphite rod anode and a platinum plate cathode. Under air, using Et4NOTs (2 equiv.) as the supporting electrolyte and MeCN/H2O (6:1) as the solvent, the desired trifluoromethyl substituted oxindole product 3a was isolated in 75% yield under constant current of 12 mA at 80 ℃ for 4 h (Table 1, entry 1). It was found that the mixed solvent of CH3CN/H2O (6:1) was better than the CH3CN and others (Table 1, entries 2–5). The temperature was found to be crucial to this reaction. Lowering the temperature led to the decrease of the yield and the product was isolated in 29% yield when the reaction was carried out at room temperature. In contrast, increasing the temperature to 100 ℃, the yield decreased to 30%, which might be ascribed to the decomposition of TfNHNHBoc at 100 ℃ (Table 1, entries 6–8, more details see Supporting information) [14b]. Furthermore, the reaction yields were decreased when the operating current was increased or decreased (Table 1, entries 9 and 10). The screening of the electrolyte showed that nBu4NClO4 or Et4NPF6 was less efficient and nBu4NI failed to offer the trifluoromethyl products (Table 1, entries 11–13). In addition, the replacement of graphite rod anode with reticulated vitreous carbon (RVC) or platinum plate led to lower yield (Table 1, entries 14 and 15). No product was observed in the absence of electricity (Table 1, entry 16).
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
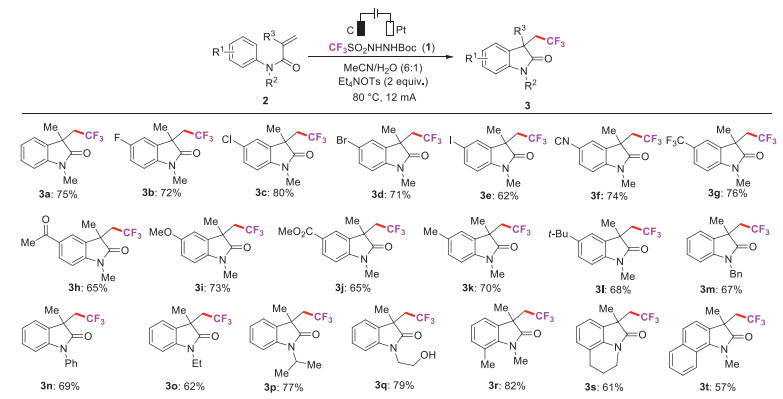
Under the above optimal reaction conditions, we investigated the scope of substrates (Scheme 2). In general, a variety of N-arylacrylamides derivatives were applicable and the corresponding trifluoromethylated oxindole products were obtained in moderate to good yields (3a-3t). Substrates with both electron-withdrawing (3b-3h, 3j) and electron-donating (3i, 3k, 3l) groups at the aryl ring reacted to give the desired products in good yields. Notably, common functional groups were well tolerated under the electrochemical conditions, such as halogen (3b-3e), trifluoromethyl (3g), cyano (3f), ester (3h), alkyl (3k, 3l) and hydroxyl groups (3q). The sterically hindered N-arylacrylamides with ortho-substituted group could also transform to the desired product in 82% yield (3r). Substrates bearing with various alkyl (3m, 3o, 3p, 3q) and aryl group (3n) at nitrogen atom were also suitable for this reaction. Gratifying, tetrahydroquinoline derivative successfully reacted with reagent 1 to afford the tricycle oxlines product (3s) in 61% yield. In addition, the reaction of N-naphthylacrylamide with reagent 1 also gave the product in 57% yield (3t).

|
Download:
|
| Scheme 2. Catalyst-free electrochemical tandem trifluoromethyl-ation/cyclization using TfNHNHBoc as a CF3 source. Reaction conditions: graphite rod (φ6 mm) anode, Pt plate (10 mm × 10 mm) cathode, 1 (0.9 mmol), 2 (0.3 mmol), electrolyte (2 equiv.), MeCN/H2O = 6:1 (5 mL); the electrolysis was conducted at a constant current in an undivided cell; under air at 80 ℃ for 4 h. Isolated yields. | |
{kind=link}
Considering the significant advantages of electro-oxidative trifluoromethylation without catalyst and oxidant, we had a strong interest in its reaction mechanism. For this purpose, we performed cyclic voltammetry (CV) experiments (Fig. 1 and Fig. S4 in Supporting information). An oxidation peak of 1 in MeCN was observed at 0.72 V (Fig. 1, curve b). At the same time, anodic oxidation of 2a, followed by an irreversible reaction, occurred at a potential of ca. 2.03 V (vs. SCE) (Fig. 1, curve c). In addition, the oxidation potential of BHT was much higher than that of TfNHNHBoc 1, ca. 1.45 V (vs. SCE) (Fig. 1, curve e), but lower than that of substrate 2a. These results indicated that reagent 1 was likely to be first oxidized under electrolytic conditions.

|
Download:
|
| Fig. 1. Cyclic voltammetry studies. (a) Cyclic voltammograms of Et4NOTs (0.1 mol/L) in acetonitrile (curve a). (b) TfNHNHBoc 1 (0.06 mol/L), Et4NOTs (0.10 mol/L) in acetonitrile (curve b). (c) 2a (0.06 mol/L), Et4NOTs (0.10 mol/L) in acetonitrile (curve c). (d) TfNHNHBoc 1 (0.18 mol/L in CH3CN), 2a (0.06 mol/L), Et4NOTs (0.10 mol/L) in acetonitrile (curve d). (e) BHT (0.06 mol/L), Et4NOTs (0.10 mol/L) in acetonitrile (curve e). Scan rate: 100 mV/s. | |
{kind=link}
To further explore the possible mechanism of the electrochemical trifluoromethylation/cyclization sequence, we carried out the control experiments. It was found that when 3.0 equiv. of radical scavengers such as BHT were added, the tandem sequences were fully suppressed. This experiment suggested that free radical intermediate was involved during the electrolysis (Scheme 3).

|
Download:
|
| Scheme 3. Radical trapping experiment. | |
{kind=link}
To evaluate the potential of this electrochemical protocol for the future application, a gram scale reaction of 2a (7 mmol, 1.2 g) with 1 was performed at a constant current of 200 mA under air. To our delight, this reaction afforded the desired product 3a with 55% yield in 5 h (Scheme 4).

|
Download:
|
| Scheme 4. Gram-scale experiment. | |
{kind=link}
Based on the above mechanism research and literature reports [21, 14a], the possible mechanism of electrochemical trifluoromethylation was proposed in Scheme 5. Reagent 1 was oxidized on the anode to generate diazene I, which decomposed to give trifluoromethyl radical (•CF3) [22]. Next, the generated CF3 radical regioselectively reacted with the alkene moiety of N-arylacrylamide 2 to give carbon center radical II, which was further converted to intermediate III through intramolecular radical cyclization. Finally, the oxidation of intermediate III and subsequent proton elimination afforded the trifluoromethylated oxindole 3. Meanwhile, protons were reduced on the cathode to produce hydrogen.

|
Download:
|
| Scheme 5. Proposed mechanism. | |
{kind=link}
In summary, we developed a new electrochemical strategy for trifluoromethylation/cyclization using TfNHNHBoc as a CF3 source under oxidant-free and catalyst-free conditions. This method provided an efficient synthesis of various trifluoromethylated oxindoles with good functional group compatibility and broad substrate scope. Preliminary mechanistic studies indicate that the reaction proceeds by a radical process. Further application of TfNHNHBoc as the CF3 source in organic electrosynthesis is currently ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Natural Science Foundation of Shanghai (No. 20ZR1471600), the Science and Technology Commission of Shanghai Municipality (No. 19DZ2271100) and the Open Research Fund Program of CAS Key Laboratory of Energy Regulation Materials (No. ORFP2020–06).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.06.008.
| [1] |
(a) E. Merino, C. Nevado, Chem. Soc. Rev. 43 (2014) 6598-6608; (b) C. Alonso, M.D.M. Eduardo, G. Rubiales, et al., Chem. Rev. 115 (2015) 1847-1935; (c) H.X. Song, Q.Y. Han, C.L. Zhao, et al., Green Chem 20 (2018) 1662-1731; (d) H. Egami, M. Sodeoka, Angew. Chem. Int. Ed. 53 (2014) 8294-8308; (e) S. Barata-Vallejo, B. Lantaño, A. Postigo, Chem. Eur. J. 20 (2014) 16806-16829. |
| [2] |
(a) O.A. Tomashenko, V.V. Grushin, Chem. Rev. 111 (2011) 4475-4521; (b) L. Chu, F.L. Qing, Acc. Chem. Res. 47 (2014) 1513-1522; (c) J. Charpentier, N. Früh, A. Togni, Chem. Rev. 115 (2015) 650-682; (d) X. Liu, C. Xu, M. Wang, et al., Chem. Rev. 115 (2015) 683-730; (e) C. Ni, M. Hu, J. Hu, Chem. Rev. 115 (2015) 765-825; (f) B. Lantaño, M.R. Torviso, S.M. Bonesi, et al., Coord. Chem. Rev. 285 (2015) 76-108. |
| [3] |
(a) H. Ge, B. Wu, Y. Liu, et al., ACS. Catal. 10 (2020) 12414-12424; (b) H. Ge, Q. Shen, Org. Chem. Front. 6 (2019) 2205-2209; (c) A. Studer, Angew. Chem. Int. Ed. 51 (2012) 8950-8958. |
| [4] |
(a) X. Mu, T. Wu, H.Y. Wang, et al., J. Am. Chem. Soc. 134 (2012) 878-881; (b) J. Li, Y. Lei, Y. Yu, et al., Org. Let. 19 (2017) 6052-6055; (c) L. Li, M. Deng, S.C. Zheng, et al., Org. Lett. 16 (2014) 504-507; (d) J.S. Lin, X.G. Liu, X.L. Zhu, et al., J. Org. Chem. 79 (2014) 7084-7092; (e) Y.F. Wang, J. Qiu, D. Kong, et al., Synlett 25 (2014) 1731-1734; (f) W. Fu, F. Xu, Y. Fu, et al., Eur. J. Org. Chem. 2014 (2014) 709-712. |
| [5] |
(a) Q. Guo, M. Wang, Y. Wang, et al., Chem. Commun. 53 (2017) 12317-12320; (b) S. Choi, Y.J. Kim, S.M. Kim, et al., Nat. Commun. 5 (2014) 4881; (c) S. Tang, Z.H. Li, M.W. Wang, et al., Org. Biomol. Chem. 13 (2015) 5285-5288; (d) Q. Li, W. Fan, D. Peng, et al., ACS. Catal. 10 (2020) 4012-4018. |
| [6] |
(a) L. Zhang, Z. Li, Z.Q. Liu, Org. Lett. 16 (2014) 3688-3691; (b) W. Wei, J. Wen, D. Yang, et al., J. Org. Chem. 79 (2014) 4225-4230; (c) R. Sakamoto, H. Kashiwagi, S. Selvakumar, et al., Org. Biomol. Chem. 14 (2016) 6417-6421. |
| [7] |
(a) D.F. Luo, J. Xu, Y. Fu, et al., Tetrahedron. Lett. 53 (2012) 2769-2772; (b) J. Xu, D.F. Luo, B. Xiao, et al., Chem. Commun. 47 (2011) 4300-4302. |
| [8] |
(a) H. Egami, R. Shimizu, M. Sodeoka, J. Fluorine. Chem. 152 (2013) 51-55; (b) P. Xu, J. Xie, Q. Xue, et al., Chem. Eur. J. 19 (2013) 14039-14042; (c) J.H. Ye, L. Zhu, S.S. Yan, et al., ACS. Catal. 7 (2017) 8324-8330; (d) L. Wu, F. Wang, X. Wan, et al., J. Am. Chem. Soc. 139 (2017) 2904-2907; (e) B. Janhsen, A. Studer, J. Org. Chem. 82 (2017) 11703-11710; (f) J.H. Ye, L. Song, W.J. Zhou, et al., Angew. Chem. Int. Ed. 128 (2016) 10176-10180; (g) F. Wang, D. Wang, X. Wan, et al., J. Am. Chem. Soc. 138 (2016) 15547-15550; (h) M. Yang, W. Wang, Y. Liu, et al., Chin. J. Chem. 32 (2014) 833-837; (i) R. Zhu, S.L. Buchwald, Angew. Chem. Int. Ed. 125 (2013) 12887-12890; (j) N.Y. Yang, Z.L. Li, L. Ye, et al., Chem. Commun. 52 (2016) 9052-9055. |
| [9] |
(a) M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230-13319; (b) K.D. Moeller, Chem. Rev. 118 (2018) 4817-4833; (c) C. Kingston, M.D. Palkowitz, Y. Takahira, et al., Acc. Chem. Res. 53 (2020) 72-83; (d) B.K. Peters, K.X. Rodriguez, S.H. Reisberg, et al., Science 363 (2019) 838-845; (e) Y. Yuan, A. Lei, Nat. Commun. 11 (2020) 802. |
| [10] |
(a) A.G. O'Brien, A. Maruyama, Y. Inokuma, et al., Angew. Chem. Int. Ed. 53 (2014) 11868-11871; (b) G.Y. Dou, Y.Y. Jiang, K. Xu, et al., Org. Chem. Front. 6 (2019) 2392-2397; (c) Y. Deng, F. Lu, S. You, et al., Chin. J. Chem. 37 (2019) 817-820; (d) C. Xu, Y. Liu, H. Liu, et al., Tetrahedron. Lett. 61 (2020) 152226. |
| [11] |
(a) K. Arai, K. Watts, T. Wirth, ChemistryOpen 3 (2014) 23-28; (b) K.Y. Ye, G. Pombar, N. Fu, et al., J. Am. Chem. Soc. 140 (2018) 2438-2441; (c) L. Zhang, G. Zhang, P. Wang, et al., Org. Lett. 20 (2018) 7396-7399; (d) W. Jud, C.O. Kappe, D. Cantillo, Chem. Eur. J. 24 (2018) 17234-17238; (e) Z. Zou, W. Zhang, Y. Wang, et al., Org. Lett. 21 (2019) 1857-1862; (f) X. Sun, H.X. Ma, T.S. Mei, et al., Org. Lett. 21 (2019) 3167-3171. |
| [12] |
(a) Z. Li, L. Jiao, Y. Sun, et al., Angew. Chem. Int. Ed. 59 (2020) 7266-7270; (b) A. Claraz, T. Courant, G. Masson, Org. Lett. 22 (2020) 1580-1584; (c) J.C. Kang, Y.Q. Tu, J.W. Dong, et al., Org. Lett. 21 (2019) 2536-2540; (d) K.Y. Ye, Z. Song, G.S. Sauer, et al., Chem. Eur. J. 24 (2018) 12274-12279; (e) G.S. Sauer, S. Lin, ACS. Catal. 8 (2018) 5175-5187. |
| [13] |
(a) J. Sun, J.K. Qiu, Y.L. Zhu, et al., J. Org. Chem. 80 (2015) 8217-8224; (b) S.R. Guo, W.M. He, J.N. Xiang, et al., Chem. Commun. 50 (2014) 8578-8581; (c) G. Rong, J. Mao, H. Yan, et al., J. Org. Chem. 80 (2015) 4697-4703; (d) T.T. Wang, F.L. Yang, S.K. Tian, Adv. Synth. Catal. 357 (2015) 928-932; (e) F.L. Yang, Y. Gui, B.K. Yu, et al., Adv. Synth. Catal. 358 (2016) 3368-3372; (f) F. Xiao, H. Xie, S. Liu, et al., Adv. Synth. Catal. 356 (2014) 364-368; (g) F.L. Yang, F.X. Wang, T.T. Wang, et al., Chem. Commun. 50 (2014) 2111-2113; (h) J. Zhang, Y. Shao, H. Wang, et al., Org. Lett. 16 (2014) 3312-3315; (i) M. Zhang, P. Xie, W. Zhao, et al., J. Org. Chem. 80 (2015) 4176-4183; (j) X. Zhao, L. Zhang, T. Li, et al., Chem. Commun. 50 (2014) 13121-13123; (k) X. Zhao, L. Zhang, X. Lu, et al., J. Org. Chem. 80 (2015) 2918-2924; (l) N. Singh, R. Singh, D.S. Raghuvanshi, et al., Org. Lett. 15 (2013) 5874-5877; (m) R. Singh, D.S. Raghuvanshi, K.N. Singh, Org. Lett. 15 (2013) 4202-4205. |
| [14] |
(a) J.Y. Guo, R.X. Wu, J.K. Jin, et al., Org. Lett. 18 (2016) 3850-3853; (b) J.Y. Guo, R.H. Dai, W.C. Xu, et al., Chem. Commun. 45 (2018) 8980-8982; (c) K. Lu, Q. Li, X. Xi, et al., Org. Chem. Front. 5 (2018) 3088-3092. |
| [15] |
(a) M. Lu, Z. Liu, J. Zhang, et al., Org. Biomol. Chem. 16 (2018) 6564-6568; (b) L. Zheng, C. Yang, Z. Xu, et al., J. Org. Chem. 80 (2015) 5730-5736; (c) L. Zhang, Z. Li, Z.Q. Liu, Org. Lett. 16 (2014) 3688-3691. |
| [16] |
(a) J. Guo, C. Xu, L. Wang, et al., Org. Biomol. Chem. 17 (2019) 4593-4599; (b) Z. Yang, A. Tang, Synlett 30 (2019) 1061-1066; (c) Z. Yang, Y. Cheng, J. Long, et al., New. J. Chem. 43 (2019) 18760-18766. |
| [17] |
Y.Y. Jiang, G.Y. Dou, K. Xu, et al., Org. Chem. Front. 5 (2018) 2573-2577. DOI:10.1039/C8QO00645H |
| [18] |
Z. Zhang, L. Zhang, Y. Cao, et al., Org. Lett. 21 (2019) 762-766. DOI:10.1021/acs.orglett.8b04010 |
| [19] |
Z. Ruan, Z. Huang, Z. Xu, et al., Org. Lett. 21 (2019) 1237-1240. DOI:10.1021/acs.orglett.9b00361 |
| [20] |
J.B. Hendrickson, D.D. Sternbach, J. Org. Chem. 40 (1975) 3450-3452. DOI:10.1021/jo00911a036 |
| [21] |
(a) P. Xiong, H.H. Xu, J. Song, et al., J. Am. Chem. Soc. 140 (2018) 2460-2464; (b) H.H. Xu, J. Song, H.C. Xu, ChemSusChem 12 (2019) 3060-3063. |
| [22] |
J.B. Tommasino, A. Brondex, M. Médebielle, et al., Synlett 2002 (2002) 1697-1699. |