 2022, Vol. 33
2022, Vol. 33 


b Qinghai Provincial Key Laboratory of Tibetan Medicine Research and CAS Key Laboratory of Tibetan Medicine Research, Northwest Institute of Plateau Biology, Xining 810008, China;
c School of Chemistry and Chemical Engineering, Shihezi University, Shihezi 832000, China
Sulfur-containing compounds are highly valuable class of organic molecules, which are widely present in functional materials, biologically active molecules and natural products [1-3]. The introduction of a sulfur-containing group into organic framework could enhance its synthetic diversity or biological activities [4-6]. Consequently, numerous efforts have been paid to construct sulfur-containing molecules by using of different sulfur-reagents including sulfonyl chlorides, sulfonylazides, sulfonyl hydrazides, sulfinate salts, thiols, S8, inorganic sulfites, and sulfur dioxide or sulfur dioxide surrogates [7-28]. Most reactions usually encounter problems with relatively complex or harsh reaction conditions and low atom efficiency. Recently, sulfinic acids as odorless, stable and readily available sulfur reagents have been increasingly employed for the synthesis of various important sulfur-containing compounds such as organic sulfones, sulfoxides, thioethers, sulfonamides and phosphorothioates with high synthetic efficiency and atom economy. During the past decade, a number of sulfonylation, sulfinylation and sulfenylation reactions using sulfinic acids have been exploited in the presence of transtion-metal/metal-free catalysis, photocatalysis or electrocatalysis. Although some great progress has been made in this field, a comprehensive review on the application of sulfinic acids to access organosulfur compounds is still lacking.
This review summarizes the recent advances in the construction of various sulfur-containing compounds using sulfinic acids as sulfur-reagents. The main achievements on this field are described in term of the reaction pattern of sulfinic acids. Selected examples of substrates are included in the text. Furthermore, specific emphasis is focused on the detailed reaction mechanism with an aim to stimulate the interest of researchers to develop more practical and versatile method by using of sulfinic acids. Finally, a personal outlook of future research will be presented in this review.
2. SulfonylationSulfone-containing molecules exhibit important functions in organic synthesis, materials science and medicinal industry [29-31]. The significance of sulfone functionalities has inspired synthetic chemists to develop new methods for their incorporation into organic moiety. Recently, sulfonyl radical-mediated procedure has been developed as a particularly useful protocol for the rapid synthesis of organic sulfones. In this context, the addition of sulfinic acids to carbon–carbon unsaturated bonds through a radical process has drawn much attention under transition-metal catalysis or metal-free conditions.
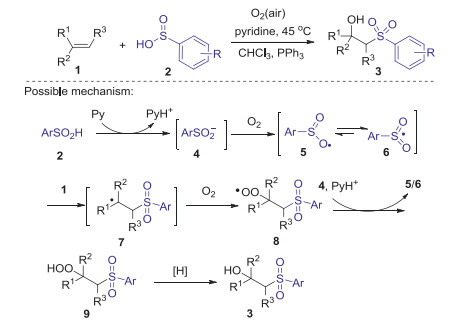
2.1. Sulfonylation of alkenesThe first example of the radical sulfonylation of alkenes using arylsulfinic acids as sulfonyl source was reported by Lei and co-workers in 2013 [32]. The procedure of this pyridine mediated oxysulfonylation of alkenes with arylsulfinic acids and dioxygen to access a series of secondary and tertiary β-hydroxysulfones with good functional-groups tolerance, in which C-S and C-O bonds were newly formed in one-pot procedure (Scheme 1). Radical trapping experiments revealed that this reaction involved a radical process. Firstly, the reaction of arylsulfinic acid with pyridine gave free sulfinyl anion 4, which was further oxidized by dioxygen to form an oxygen centered radical 5 via single electron transfer process. The resonance of oxygen centered radical 5 generated sulfonyl radical 6. Subsequently, the radical addition of 6 to alkene 1 produced alkyl radical 7, which reacted with dioxygen to afford alkylhydroperoxy radical intermediate 8 through a redox-transfer process. Next, β-peroxylsulfone 9 was generated from the intermediate 8 via single-electron transfer (SET) and along with proton transfer from 4 and pyridium. Finally, β-peroxylsulfone 9 was reduced by sulfinic acid or PPh3 to form the desired β-hydroxysulfones 3.

|
Download:
|
| Scheme 1. Aerobic oxysulfonylation of alkenes with arylsulfinic acids and dioxygen. | |
{kind=link}
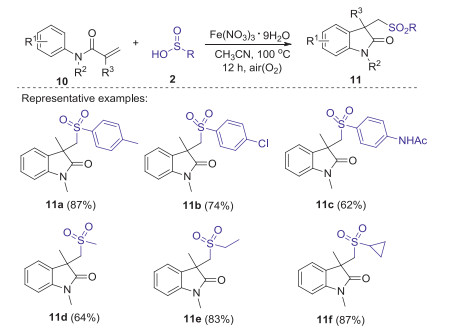
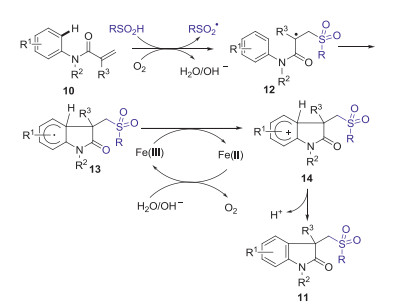
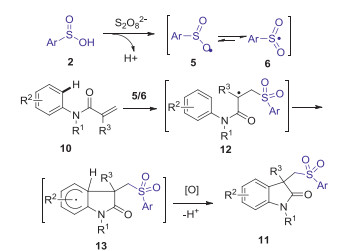
Substituted oxindoles display diverse fascinating biological and pharmacological activities [33]. In 2014, Jiao group reported Fe-catalyzed aerobic oxidative sulfonyl-carbocyclization of activated alkenes with sulfinic acids for the synthesis of sulfonyl substituted oxindoles [34]. Dioxygen in air was used as a green oxidant and played a key role in initiating this procedure. A series of substituted arylsulfinic acids with different substituents (Me, Cl, NHAc) and alkyl sulfinic acids (methyl, ethyl, cyclopropyl) could smoothly produce desired sulfonated oxindoles in moderate to excellent yields under the standard conditions (Scheme 2). A possible mechanism is demonstrated as shown in Scheme 3. Initially, the oxidation of sulfinic acids by O2 in air generated the sulfonyl radical. Subsequently, the addition of sulfonyl radical to activated alkene 10 gave radical intermediate 12. Then, the intramolecular cyclization of intermediate 12 gave radical intermediate 13, which is further oxidized by Fe(Ⅲ) to generate cationic intermediate 14. Finally, the deprotonation of intermediate 14 produced desired sulfonyl substituted oxindoles 11.

|
Download:
|
| Scheme 2. Fe-catalyzed aerobic sulfonyl-carbocyclization of activated alkenes. | |
{kind=link}

|
Download:
|
| Scheme 3. Possible mechanism for Fe-catalyzed aerobic sulfonyl-carbocyclization of activated alkenes. | |
{kind=link}
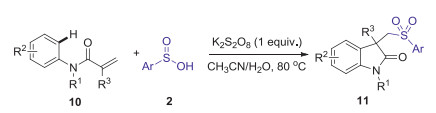
The same year, Wei and Wang also reported a metal-free direct arylsulfonylation of arylacrylamides with sulfinic acids to access a diverse range of sulfonated oxindoles by simply using the cheap K2S2O8 as the oxidant (Scheme 4) [35]. A radical mechanism was proposed as shown in Scheme 5.

|
Download:
|
| Scheme 4. Metal-free direct arylsulfonylation of arylacrylamides with sulfinic acids. | |
{kind=link}

|
Download:
|
| Scheme 5. Possible mechanism for metal-free direct arylsulfonylation of arylacrylamides with sulfinic acids. | |
{kind=link}
In 2014, Wei and Wang reported a novel iron-catalyzed difunctionalization of alkenes with sulfinic acids and dioxygen for the construction of β-ketosulfones [36]. This protocol provides a simple, convenient and environmentally benign approach to access a series of β-ketosulfones, which has the advantages of inexpensive catalyst, readily available sulfonylating reagents, green oxidant and oxygen source. According to the proposed mechanism, the sulfonyl radical 6 is firstly produced from sulfinic acids 2 via the single electron transfer (SET) and deprotonation process with the aid of iron salt and dioxgyen. Then, the addition of sulfonyl radical 6 to alkene 1 produced the alkyl radical 16. Next, the interaction of alkyl radical 16 with dioxygen leading to peroxy radical 17, which interacted with ·OOH to generate monoalkyl tetroxide 18. Finally, the decomposition of intermediate 18 produced β-ketosulfones 15 (Scheme 6).

|
Download:
|
| Scheme 6. Iron-catalyzed difunctionalization of alkenes with sulfinic acids and dioxygen for the construction of β-ketosulfone. | |
{kind=link}
In 2015, Lei's group described a CuBr2 catalyzed oxysulfonylation of arylacrylic acids with sulfinic acids and dioxygen leading to β-ketosulfones [37]. Various substituted β-ketosulfones could be efficiently obtained through a sequence of S–H bond alkylation, C–C σ bond cleavage, and aerobic oxygenation process (Scheme 7). Electron paramagnetic resonance spectroscopies and operando X-ray absorption offer a clear evidence for the single electron redox process between Cu(Ⅱ) and sulfinic acids.

|
Download:
|
| Scheme 7. CuBr2-catalyzed oxysulfonylation of arylacrylic acids with sulfinic acids and dioxygen. | |
{kind=link}
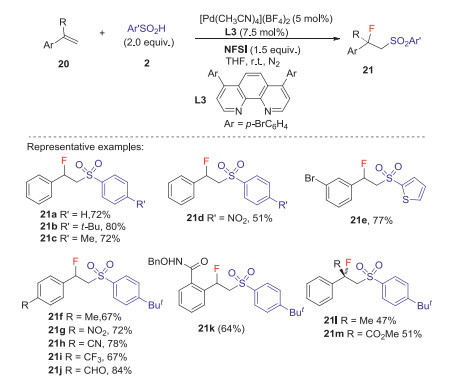
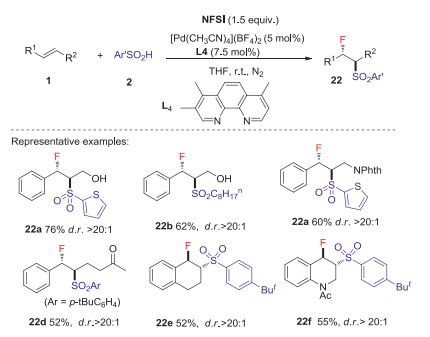
The same year, Liu group [38] reported the first Pd catalyzed intermolecular anti-specific fluorosulfonylation of alkenes with arylsulfinic acids and NFSI to deliver β-fluoro sulfones with excellent regio- and diastereoselectivity, in which phenanthroline type ligands L3 and L4 were used as the key additives. This reaction exhibits excellent functional-group tolerance in various arylsulfinic acids and alkenes to provide a series of β-fluoro sulfones in moderate to good yields (Schemes 8 and 9). Preliminary mechanistic study revealed that a high-valent L2PdⅢF species was involved in the C-F bond formation. The detailed mechanism was shown in Scheme 10. Initially, [(L4)2Pd(F)N(SO2Ph)2]2+ 24 was formed through the oxidation of cationic [(L4)2Pd]2+ 23 by NFSI. The reaction of active catalyst 24 with arylsulfinic acid to generate the ArSO2 radical 6 and (L4)2PdⅢF species via an SET process. The sulfonyl radical could react with alkene 1 to give carbon radical 25, which reacted with PdⅢF complex to deliver the β-fluoro sulfone 22 (dash line). Alternatively, the alkyl radical 25 could also be attacked by the PdⅢF complex to produce the alkyl-PdⅣF species 26, which underwent reductive elimination process to give the desired product (plain cycle).

|
Download:
|
| Scheme 8. Fluorosulfonylation of styrenes with arylsulfinic acids. | |
{kind=link}

|
Download:
|
| Scheme 9. Fluorosulfonylation of internal alkenes. | |
{kind=link}

|
Download:
|
| Scheme 10. Possible mechanism for Pd-catalyzed intermolecular fluorosulfonylation of styrenes. | |
{kind=link}
In 2016, the Lei group [39] reported visible-light induced sulfonylation of α-methyl-styrene derivatives with sulfinic acids leading to allylic sulfones, in which Eosin Y and Co(dmgH)2pyCl were employed as a co-photocatalyst. The reaction exhibited a broad substrate scope and the corresponding products were obtained in moderate to good yields. Based on the experimental results and their previous reports, a possible mechanism was proposed in Scheme 11. Initially, arylsulfinic acid reacted with pyridine produced sulfinyl anion 4, which was oxidized by excited Eosin Y* to give an oxygen centered radical 5 through single electron transfer under visible light irradiation. The resonance of oxygen radical 5 formed sulfonyl radical 6, which added to α-methylstyrene 20 to generate alkyl radical 28. Subsequently, alkyl radical 28 is oxidized by Co(dmgH)2pyCl to deliver a Co(Ⅱ) species and a cation intermediate 29. Finally, the elimination of hydrogen ion from intermediate 29 afforded desired allylic sulfone 27. Meanwhile, the single-electron reduction of Co(Ⅱ) species by the Eosin Y radical anion produced a Co(Ⅰ) species and completed the photocatalytic cycle.

|
Download:
|
| Scheme 11. Visible-light induced sulfonylation of α-methyl-styrene derivatives with sulfinic acids leading to allylic sulfone. | |
{kind=link}
Afterwards, Yang and Wang developed visible-light mediated and Eosin Y catalyzed method for the construction of β-ketosulfones from alkenes and sulfinic acids in the presence of TBHP under the irradiation of 11 W white LED [40]. A number of aromatic alkenes bearing either electron-donating groups (R = OMe, Me) or electron-withdrawing groups (R = Cl, Br, CN, NO2) on the aryl ring, reacted smoothly with sulfinic acids, providing the desired β-ketosulfones. Nevertheless, alkyl alkenes was not suitable for this reaction system. The mechanism studies suggested that a radical process should be involved in this transformation and the carbonyl oxygen atom of the β-ketosulfones came from both TBHP and H2O (Scheme 12).

|
Download:
|
| Scheme 12. Visible-light mediated synthesis of β-ketosulfones from alkenes and sulfinic acids. | |
{kind=link}
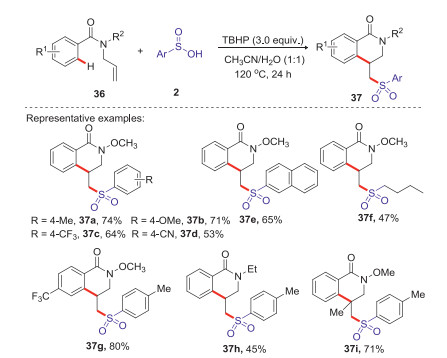
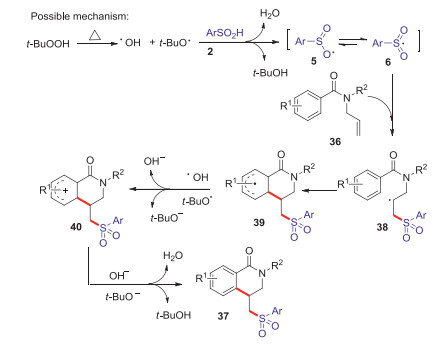
Dihydro isoquinolones are widely existed in various natural products and drug molecules, which exhibited a wide range of interesting biological properties such as anti-inflammatory, anti-allergic and anti-tumor [41]. In 2016, Wang group reported an efficient strategy for the synthesis of sulfonated dihydroisoquinolinones via TBHP mediated arylsulfonylation reaction of N-allylbenzamides with arylsulfinic acids at 120 ℃ (Scheme 13) [42]. Various sulfonated dihydroisoquinolones were conveniently obtained in moderate to good yields through radical addition and intramolecular cyclization process. A plausible mechanism is shown in Scheme 14. Firstly, hydroxyl and tert-butoxyl radicals were formed through the homolysis of TBHP under heating conditions. The abstraction of hydrogen from arylsulfinic acids generated an oxygen centered radical 5 resonating with the sulfonyl radical 6. Subsequently, the addition of sulfonyl radical 6 to the activated alkene 1 giving the alkyl radical intermediate 38, which underwent the intramolecular radical cyclization to afford a radical intermediate 39. Then, the radical intermediate 39 was oxidized by the tert-butoxyl radical or hydroxyl radical to afford a cationic intermediate 40 via a SET process. Finally, the lose of proton from the cationic intermediate 40 to produce the desired sulfonated isoquinolinone 37 and with the release of water or tert-butanol.

|
Download:
|
| Scheme 13. TBHP mediated arylsulfonylation reaction of N-allylbenzamides with arylsulfinic acids leading to sulfonated dihydroisoquinolinones. | |
{kind=link}

|
Download:
|
| Scheme 14. Possible mechanism for the synthesis of sulfonated dihydroisoquinolinones. | |
{kind=link}
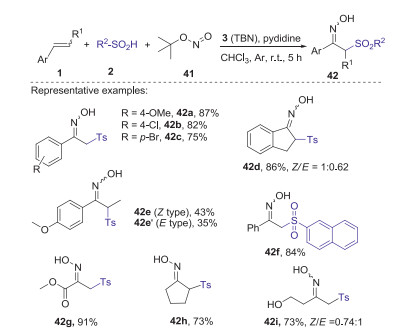
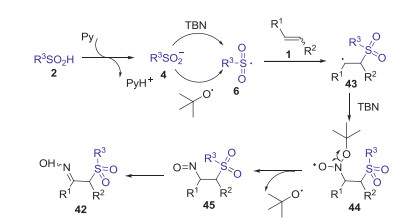
In 2017, Yu and Han group presented a novel TBN-mediated method for synthesis of α-sulfonylketoximes via the sulfoximation of alkenes with sulfinic acids and tert-butyl nitrite (TBN) [43]. This strategy has broad substrate scope and good reaction efficiency, in which both aromatic olefins and unactivated aliphatic alkenes were all well compatible with this procedure (Scheme 15). Proposed mechanism is showed in Scheme 16. Firstly, sulfinic acid 2 was deprotonated by pyridine to form sulfinyl anion 4, which was further oxidized by TBN to access sulfonyl radical 6 via single electron transfer (SET) process. Subsequently, the addition of sulfonyl radical 6 to alkene 1 gave the alkyl radical 43, which was trapped by TBN affording intermediate 44. Next, the elimination of a tert-butoxyl radical (t-BuO·) from intermediate 44 provided nitroso compound 45. Finally, the tautomerization of nitroso compound 45 produced product 42.

|
Download:
|
| Scheme 15. TBN-mediated vicinal sulfoximation of alkenes with sulfinic acids leading to α-sulfonylketoximes. | |
{kind=link}

|
Download:
|
| Scheme 16. Possible mechanism for the synthesis of α-sulfonylketoximes. | |
{kind=link}
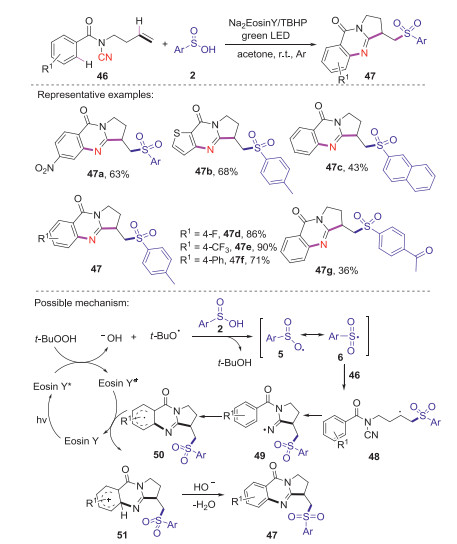
The same year, Han and co-workers reported a facile photocatalytic oxidative cyclization reaction of N-cyanamide alkenes with sulfinic acids for the synthesis of sulfonated quinazolinones [44]. The reaction could be efficiently performed by using of Na2-Eosin Y as photocatalyst and TBHP as the base under green light irradiation. This strategy has the advantages of readily available sulfonylation reagents, mild conditions, and broad substrate scope. The corresponding mechanism is proposed as shown in Scheme 17. Initially, the excited-state Eosin Y* was formed from Eosin Y by the irradiation of green LED light. The single electron transfer from Eosin Y* to TBHP gave tert-butyloxy radical and OH-. Subsequently, arylsulfonyl radical 6 was formed through the interaction of arylsulfinic acid with tert-butyloxy radical. Next, the sulfonyl radical 6 added to the C=C bond of N-cyanamide alkenes 46 affording the alkyl radical intermediate 48, which reacted with the cyano group leading to the nitrogen-centered radical 49. Then, intramolecular cyclization would lead to the formation of the key intermediate 50, which was further oxidized by Eosin Y*+ to give cationic intermediate 51 with the release of the photocatalyst. Finally, the deprontonation of cationic intermediate 51 produced the desired product 47.

|
Download:
|
| Scheme 17. Photocatalytic oxidative cyclization reaction of N-cyanamide alkenes with sulfinic acids for synthesis sulfonated quinazolinones. | |
{kind=link}
In 2017, Jiang and Tu reported a new visible-light mediated and Eosin Y-catalyzed arylsulfonylation and bicyclizations of C(sp3)-tethered 1, 7-enynes with sulfinic acids in the presence of K2CO3 [45]. A series of structural diverse sulfone-containing benzo[a]fluoren-5-ones could be obtained with good yields under light irradiation of 12 W blue LEDs (Scheme 18). This cascade cyclization reactions feature high reaction efficiency, broad substrates scope, excellent functional-groups tolerance, and mild reaction conditions, enabling sulfonyl radical-triggered multiple bond forming events including C-S and C-C bonds to synthesize polycyclic-linked alkyl arylsulfones.

|
Download:
|
| Scheme 18. Visible-light mediated and Eosin Y-catalyzed arylsulfonylation and bicyclizations of C(sp3)-tethered 1, 7-enynes with sulfinic acids. | |
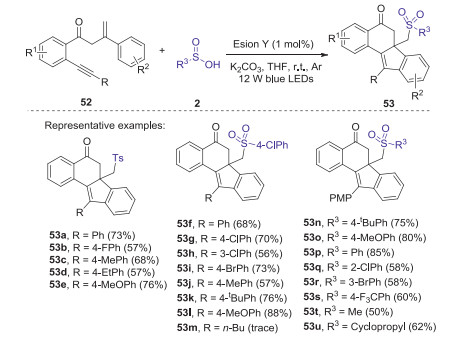{kind=link}
In 2018, Jiang and co-workers described Pd(Ⅱ)-catalyzed selective aminosulfonylation and fluorosulfonylation of carbonyl-tethered 1, 7-enynes with N-fluorobenzenesulfonimide (NFSI) and sulfinic acids (Scheme 19). A variety of functionalized (E)-3, 4-dihydronaphthalen-1(2H)-ones could be obtained in good yields with high stereoselectivity under mild and redox neutral conditions [46].

|
Download:
|
| Scheme 19. Pd(Ⅱ)-catalyzed aminosulfonylation and fluorosulfonylation of carbonyl-tethered 1, 7-enynes with N-fluorobenzenesulfonimide (NFSI) and sulfinic acids. | |
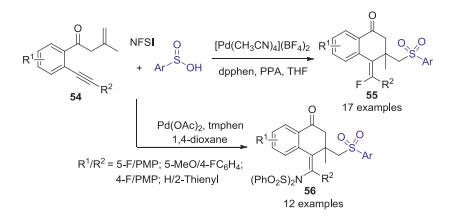{kind=link}
The same year, Jiang also reported a TBHP mediated selenosulfonylation of β-alkynyl propenones with sulfinic acids and diphenyl diselenide (Scheme 20) [47]. In this transformation, three new bonds including the C-S, C-C and C-Se bonds were directly formed under the mild oxidative conditions. This protocol provided a metal-free and convenient approach to access a range of richly decorated (Z)-1-indenones. The proposed mechanism is demonstrated as shown in Scheme 20. Firstly, sulfinic acid 2 was oxidized by TBHP to generate the aryl sulfonyl radical 6. Subsequently, aryl sulfonyl radical 6 added into β-alkynyl propenone 57 leading to intermediates 60, which underwent the 5-exo-dig cyclization to give vinyl radical 61. Finally, the coupling of vinyl radical with diphenyl diselenide afforded the desired product 59.

|
Download:
|
| Scheme 20. TBHP mediated selenosulfonylation of β-alkynyl propenones with sulfinic acids and diphenyl diselenide. | |
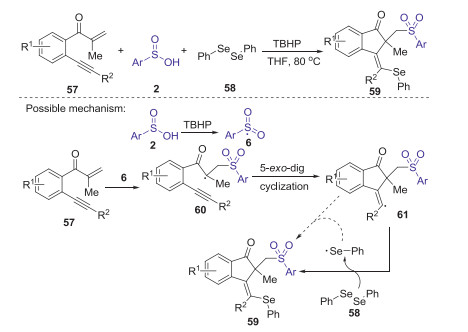{kind=link}
Functionality migration has been regarded as an efficient way to construct structurally unique and invaluable functionalized compounds in synthetic chemistry [48]. In 2018, a convenient electrooxidative sulfonylation/heteroarylation reaction of unactivated alkenes with sulfinic acids was reported by Guo and Li [49]. This electro-synthetic strategy allowing distal heteroaryl ipso-migration and the direct construction of C-S and C-C bond, provides an efficient and green approach to prepare a number of sulfone-containing molecules under an undivided cell. The mechanism studies suggested that the sulfinic acid might undergo a deprotonation process, resulting in sulfonyl radicals. The detailed mechanism has been described in Scheme 21. The sulfonyl ion 4 was firstly formed through the deprotonation of sulfinic acid 2. Then, ion intermediate 4 is oxidized to generate oxygen centered radical 5 resonating with the sulfonyl radical 6. Next, the intermolecular radical addition of sulfonyl radical 6 to benzothiazole substituted tertiary alcohols to give intermediate 64, which would lead to the formation of spiro N-radical 65 via a five-membered cyclic radical transition state. Subsequently, C-C bond cleavage and ring opening of the spirostructure occurred to give a ketyl radical 66, which was further oxidized at the anode to afford the cationic intermediate 67. Finally, the desired product 63 was produced by the deprotonation of cationic intermediate 67.

|
Download:
|
| Scheme 21. Electrooxidative sulfonylation/heteroarylation reaction of unactivated alkenes with sulfinic acids. | |
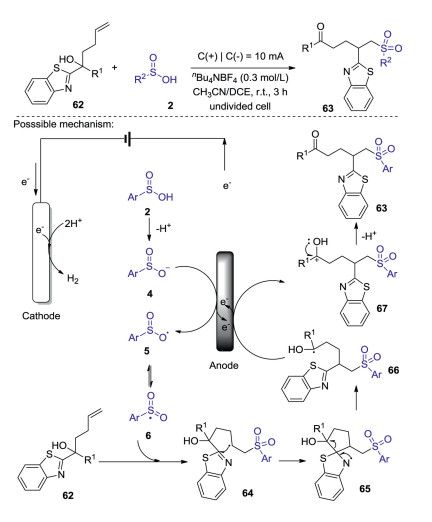{kind=link}
C3-Substituted quinoxalin-2(1H)-ones are a important class of biologically active molecules. In 2019, Koley and co-workers described a TBHP mediated three-component reaction of quinoxalin-2(1H)-ones, alkenes, and sulfinic acids for the synthesis of C3-alkyl substituted quinoxalin-2(1H)-ones [50]. This reaction underwent through a radical cascade process, which was triggered by the sulfonyl radical generated from sulfinic acid. Under the optimized reaction conditions, a range of C3 substituted quinoxalin-2(1H)-ones bearing sulfone groups are obtained in good to excellent yields under mild conditions (Scheme 22).

|
Download:
|
| Scheme 22. Three-component reaction of quinoxalin-2(1H)-ones, alkenes, and sulfinic acids for the synthesis of C3-alkyl quinoxalin-2(1H)-ones. | |
{kind=link}
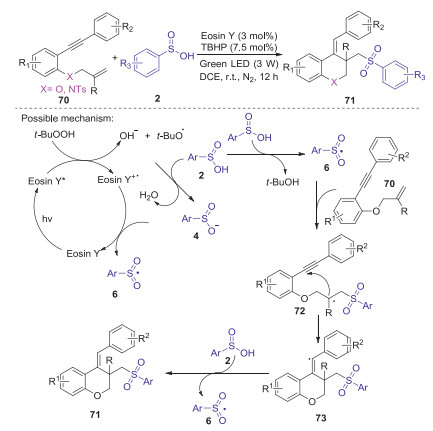
In 2020, Li and Wang's group reported a visible-light-promoted method for the construction of sulfonated chromanes and sulfonated 1, 2, 3, 4-tetrahydroquinolines through a cyclization of 1-(arylethynyl)-2-(vinyloxy)benzenes and N-allyl-2-(arylethynyl)anilines with sulfinic acids [51]. This protocol using Eosin Y (3.0 mol%) as a photocatalyst and TBHP (7.5 mol%) as an oxidant, which provides a mild and efficient approach to access various sulfonated products in good yields. A possible reaction mechanism was demonstrated in Scheme 23. Firstly, the excited-state Eosin Y* is generated from Eosin Y under visible-light irradiation, which interacted with TBHP to give HO− and t-BuO·along with the formation of Eosin Y+·. Subsequently, the abstraction of a hydrogen from arylsulfinic acid 2 by t-BuO· produced sulfonyl radical 6. Then, radical 6 added to carbon-carbon double bond of 70 to form an alkyl radical 72, which underwent the intramolecular cyclization with carbon-carbon triple bond of alkyne through a 6-exo-dig cyclization affording vinyl intermediate 73. Finally, the hydrogen transfer from 2 to intermediate 73 produced the product 71 along with the release of sulfonyl radical 6 for next cycle.

|
Download:
|
| Scheme 23. Visible-light-promoted method for the construction of sulfonated chromanes and sulfonated 1, 2, 3, 4-tetrahydroquinolines. | |
{kind=link}
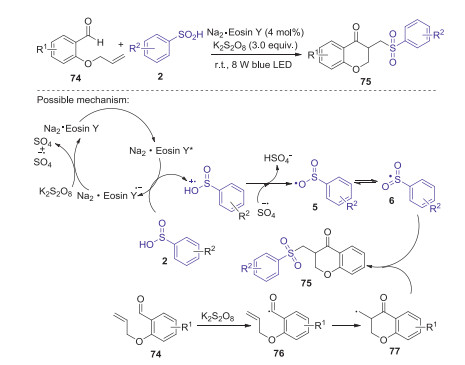
At the same time, an efficient visible-light induced cascade radical cyclization of sulfinic acids and o-(allyloxy)arylaldehydes towards functionalized chroman-4-ones was achieved by Wang and co-workers [52]. Radical reaction mechanism was proposed for this transformation. As shown in Scheme 24, firstly, sulfinic acid was oxidized by the excited Na2·Eosin Y* to generate the sulfonyl radical cation under visible-light irradiation. Subsequently, sulfonyl radical cation was deprotonated by SO4·- to give sulfonyl radical 6. Meanwhile, o-(allyloxy)arylaldehydes 74 could be oxidized by K2S2O8 to access acyl radical 76 via a single-electron transfer (SET) process. Next, 76 underwent radical cyclization reaction to afford radical 77. Finally, the coupling of intermediate 77 with sulfonyl radical 6 would lead to the formation of the desired product 75.

|
Download:
|
| Scheme 24. Visible-light induced cascade radical cyclization of sulfinic acids and o-(allyloxy)arylaldehydes. | |
{kind=link}
Chiral sulfones are highly valuable structural motifs in modern synthetic chemistry. In particular, optical pure allylic sulfones are one of the most important class of chiral sulfone building blocks owing to the versatile synthetic applications of alkene. In 2020, Zi and co-workers reported (R)-DTBM-Segphos/Pd-catalyzed regio- and enantioselective hydro-sulfonylation of 1, 3-dienes with sulfinic acids leading to 1, 3-disubstituted chiral allylic sulfones [53]. A variety of 1, 3-unsymmetrical chiral allylic sulfones that are otherwise difficult to obtain via previous protocols were efficiently constructed using this strategy, which could undergo in a step and atom-economical fashion under mild conditions (Scheme 25). Interestingly, the Pd-hydride was not involved in this reaction system. Combined computational and experimental studies indicated that this transformation was triggered by a ligand-to-ligand hydrogen transfer followed by a C-S bond reductive elimination process through a six-membered transition state.

|
Download:
|
| Scheme 25. (R)-DTBM-Segphos/Pd-catalyzed regio- and enantioselective hydro-sulfonylation of 1, 3-dienes with sulfinic acids. | |
{kind=link}
2.2. Sulfonylation of alkynes
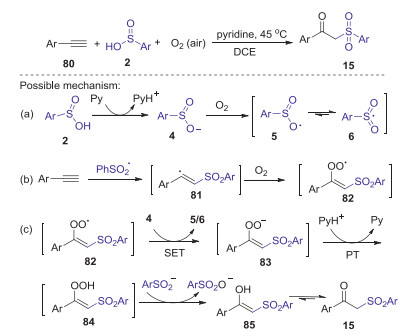
Alkynes are highly valuable building blocks, which have been increasingly utilized in various sulfonylations by using of sulfininc acids as sulfonylating agents. In 2013, a first example of dioxygen-triggered oxidative difunctionalization of terminal alkynes with arylsulfininc acids and dioxygen toward β-keto sulfones was reported by Lei group [54]. The reaction was conducted under mild conditions with high selectivity, in which pyridine not only used as a base, but also played a vital role in reduction of sulfinic acids. The detailed reaction mechanism was showed in Scheme 26. Firstly, arylsulfinyl anion 4 was generated from arylsulfinic acid in the presence of pyridine. Then, the oxidation of 4 by dioxygen produced an oxygencentered radical 5, which could be transformed into sulfonyl radical 6 via resonation process. Subsequently, the addition sulfonyl radical to alkynes gave the reactive vinyl radical 81, which could be trapped by dioxygen forming peroxide radical 82. Next, the intermediate 82 underwent via the single electron transfer and protonation process successively with 4 and pyridinium, generating 5/6 and affording peroxide 84, which was reduced by benzenesulfinic acid produced intermediate 85. Finally, the isomerization of intermediate 85 would lead to the formation of product 15.

|
Download:
|
| Scheme 26. Oxidative difunctionalization of terminal alkynes with arylsulfininc acids and dioxygen toward β-keto sulfones. | |
{kind=link}
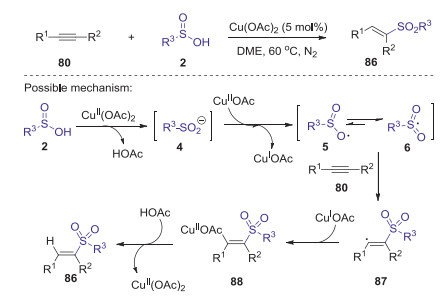
In 2014, Wei and Wang developed a novel copper catalyzed direct hydrosulfonylation of alkynes with arylsulfinic acids for the synthesis of (E)-vinyl sulfones under N2 [55]. Various arylalkynes and arylsulfinic acids bearing electron withdrawing or donating groups were all suitable substrates, affording the corresponding vinyl sulfones in moderate to good yields. It should be noted that this reaction could also be scaled up to gram-scale without any significant loss of its efficiency. Proposed mechanism is showed in Scheme 27. Firstly, sulfinic acid 2 reacted with Cu(OAc)2 generated the sulfinyl anion 4, which was oxidized by CuⅡ to afford an oxygen centered radical 5 resonating with sulfonyl radical 6. Subsequently, the selective addition of sulfonyl radical 6 to alkyne 80 afforded vinyl radical 87. Then, vinyl copper(Ⅱ) complexes 88 was formed through vinyl radical 87 interacted with CuI species. Finally, the desired product 86 was produced by the protonation of 88 with the release of Cu(Ⅱ) catalyst.

|
Download:
|
| Scheme 27. Copper catalyzed direct hydrosulfonylation of alkynes with arylsulfinic acids for the synthesis of (E)-vinyl sulfones. | |
{kind=link}
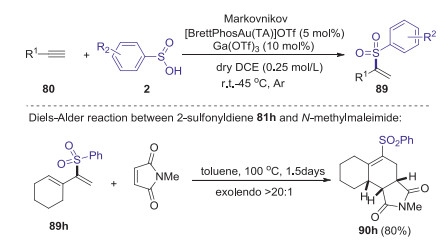
The same year, Shi group developed a gold(Ⅰ)-catalyzed method for the synthesis of α-substituted vinyl sulfones from simple terminal alkynes and sulfinic acids [56]. This homogenous gold catalysis, which utilized [BrettPhosAu(TA)]OTf (TA = 1H-benzotriazole) as catalyst and Ga(OTf)3 as additive, provide an efficient approach to construct various α-substituted vinyl sulfones in good yields with excellent selectivity (Scheme 28). The synthetic utility of α-substituted vinyl sulfones was investigated to construct tricyclic ring system with excellent endoselectivity via Michael addition of 89h with N-methylmaleimide.

|
Download:
|
| Scheme 28. Gold(Ⅰ)-catalyzed method for the synthesis of α-substituted vinyl sulfones. | |
{kind=link}
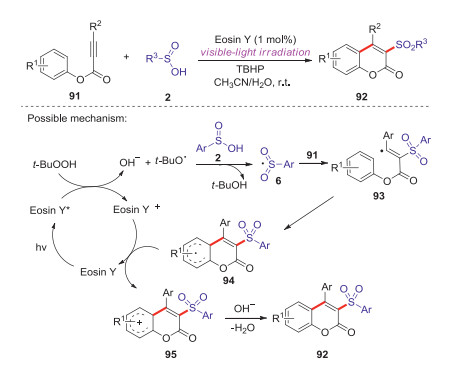
In 2015, Wang group developed a metal-free visible-light initiated approach to access 3-sulfonated coumarins via Eosin Y-catalyzed arylsulfonylation of alkynes with arylsulfinic acids [57]. This methodology provided a series of 3-sulfonated coumarins in good yields through a tandem reaction process under mild conditions. A possible mechanism for this reaction is proposed in Scheme 29. Initially, the interaction of tert-butyl hydroperoxide (TBHP) with excited state of Eosin Y* produced a tert-butoxyl radical under visible-light irradiation. Secondly, an abstraction of hydrogen radical from sulfinic acid 2 gave the corresponding sulfonyl radical 6 and t-BuOH. Then, the addition of sulfonyl radical 6 to alkyne 91 delivered the vinyl radical intermediate 93. Intramolecular cyclization of vinyl radical 93 with an arylring generated the radical intermediate 94, which was further oxidized by Eosin Y+ to give carbocation intermediate 95. Finally, the deprotonation of intermediate 95 afforded the desired 3-sulfonated coumarin product 92.

|
Download:
|
| Scheme 29. Visible-light initiated arylsulfonylation of alkynes with arylsulfinic acids. | |
{kind=link}
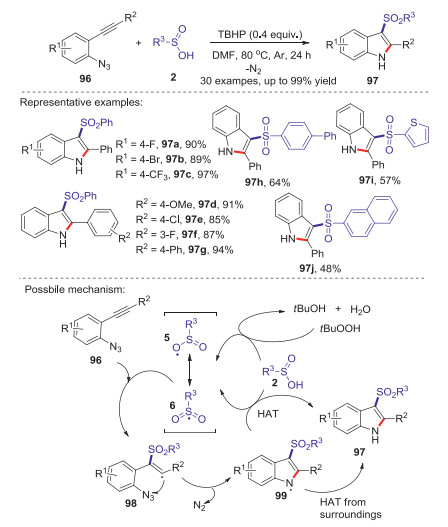
Indole frameworks are important in medicinal and biological chemistry. In 2016, Han group reported a facile and efficient TBHP mediated vicinal sulfonamination of 2-alkynyl arylazides with sulfinic acids to access 3-sulfonylindoles [58]. Through this protocol, a variety of potentially bioactive 3-sulfonylindoles were facilely synthesized in one-pot procedure by using of sulfinic acids as the sulfonating reagent and azido as the aminating reagent. The control experiments confirmed that the reaction went through a radical process. The reaction mechanism is showed in Scheme 30. Initially, the oxidation of sulfinic acid 2 by TBHP to form the radical 6. Subsequently, the vinyl intermediate 98 was generated through the addition of sulfonyl radical to the alkynyl moiety of 96. Next, intermediate 98 underwent an intramolecular cyclization of the alkenyl radical with an azido moiety to produce the N-radical intermediate 99 along with the release of N2. Finally, the transformation of hydrogen atom from sulfinic acid 2 to intermediate 99 or the surroundings produced 3-sulfonylindole.

|
Download:
|
| Scheme 30. TBHP mediated vicinal sulfonamination of 2-alkynyl arylazides with sulfinic acids to access 3-sulfonylindoles. | |
{kind=link}
The same year, Zhu group described a TBHP-initiated cyclization of oazidoaryl acetylenic ketones with sulfinic acids to construct various 3-sulfonated 4-quinolones (Scheme 31) [59]. This reaction is characterized by good functional-group tolerance, mild conditions, and ability to gram-scale synthesis. This reaction might involve two possible mechanisms: (a) radical chain propagation pathway; (b) radical-radical coupling pathway. In path a (propagation step), sulfonyl radical 6 was generated through the reaction of sulfinic acid 2 with TBHP under heating conditions. Subsequently, the sulfonyl radical 6 added to the alkynyl moiety of 100 to give the vinyl radical 102, which underwent intramolecular cyclization to produce the N-radical intermediate 103. Finally, the hydrogen abstraction from sulfinic acid to the N-radical 103 afforded the desired product 101. On the other hand, the radical-radical coupling pathway (path b) may also be involved in this transformation. Firstly, nitrene intermediate 104 formed from substrate 100 by releasing of N2 under heating condition. Then, the interaction of nitrene intermediate 104 with tert-butoxy radical to give the intermediate 105, which underwent intramolecular cyclization to afford the intermediate 106. Finally, the cross-coupling of sulfonyl radical 6 with intermediate 106 formed the intermediate 107, which would be transformed into product 101 via hydrolyzation process.

|
Download:
|
| Scheme 31. TBHP-initiated cyclization of oazidoaryl acetylenic ketones with sulfinic acids to construct various 3-sulfonated 4-quinolones. | |
{kind=link}
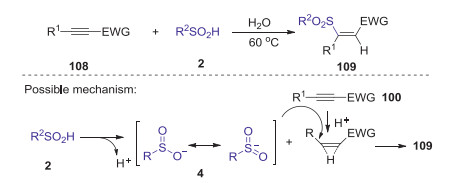
In 2016, He and co-workers reported a catalyst-free strategy for the synthesis of Z-β-sulfonyl-a, β-unsaturated carbonyl compounds through hydrosulfonation of alkynylcarbonyl compounds with sulfinic acid in water [60]. Sulfinic acid played three roles in this transformation, which was employed as hydrogen source, sulfonation reagent, and activating reagent (Scheme 32). This reaction has the excellent a good functional-group tolerance owing to its weak acidity and redox-neutral conditions. The mechanism study revealed that the addition of sulfonyl anion to ethenium intermediate was involved in this transformation.

|
Download:
|
| Scheme 32. Catalyst-free strategy for the synthesis of Z-β-sulfonyl-a, β-unsaturated carbonyl compounds. | |
{kind=link}
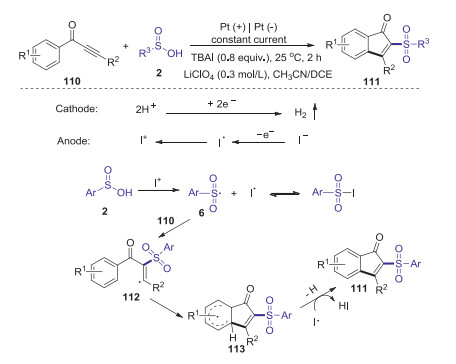
An electrooxidative direct arylsulfonlylation of ynones with sulfinic acids for the synthesis of sulfonated indenones was developed by Lei in 2017 [61]. This protocol provides a facile and efficient approach to access a series of biologically important sulfone containing indenones under constant current conditions in a simple undivided cell. A plausible reaction pathway is illustrated in Scheme 33. Firstly, the anodic oxidation of the iodide ion to form I+ species, which reacted with sulfinic acid 2 to give sulfonyl radical 6 and iodine radical. Subsequently, the addition of sulfonyl radical 6 to ynones 110 afforded vinyl radical 112. Then, the intramolecular cyclization of intermediate 112 provided the radical intermediate 113. Finally, the oxidation of 113 produced the corresponding sulfonated indenone 111.

|
Download:
|
| Scheme 33. Electrooxidative arylsulfonlylation of ynones with sulfinic acids for the syntheis of sulfonated indenones. | |
{kind=link}
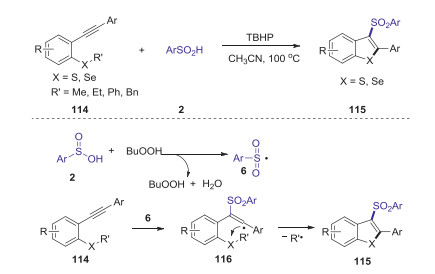
In 2017, Song group reported tert-butylhydroperoxide-initiated radical cyclization of 2-alkynylthioanisoles or -selenoanisoles with sulfinic acids leading to 3-(arylsulfonyl)benzothiophenes or -benzoselenophenes [62]. This reaction could provide the corresponding products in moderate to good yields under mild conditions, in which cascade C(sp3)-S(Se) bond cleavage and two C(sp2)-S(Se) bond formation was involved in one-pot procedure. A possible mechanism for this radical mediated cyclization reaction is presented in Scheme 34. Initially, the interaction of TBHP and sulfinic acid 2 would lead to the formation of sulfonyl radical 6. Subsequently, sulfonyl radical 6 selectively attacked the C-C triple bond of 114 to generate the vinyl radical 116, which reacted with SR' moiety gave the final product 115 through 5-exo-trig cyclization mode along with the release of alkyl radical.

|
Download:
|
| Scheme 34. tert-Butyl hydroperoxide-initiated radical cyclization of 2-alkynylthioanisoles or -selenoanisoles with sulfinic-acids. | |
{kind=link}
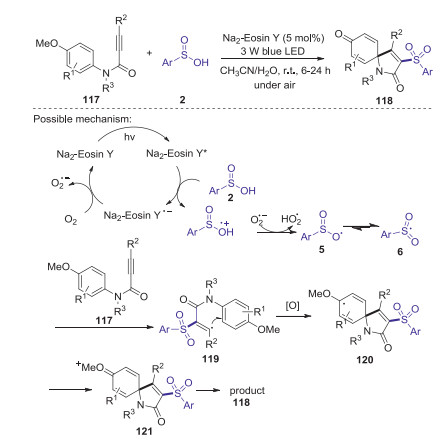
The same year, Wei and Wang reported visible-light-induced difunctionalization of activated alkynes with sulfinic acids for the construction of 3-sulfonyl azaspiro[4,5]trienones at room temperature [63]. This strategy simply utilizes visible light as green energy source and Na2-Eosin Y as inexpensive photocatalyst, providing an efficient approach to construct various 3-sulfonyl azaspiro[4,5]trienones in moderate to good yields. A possible mechanism as shown in Scheme 35. Initially, Na2-Eosin Y* was formed from Na2-Eosin Y under blue LED light-irradiation. Then, the sulfinic acid radical cation was generated through a single electron transfer process from sulfinic acid 2 to Na2-Eosin Y*. Subsequently, the deprotonation of radical cation 6 by O2·- produced the oxygen-centered radical 5 resonating with the sulfonyl radical 6. Then, the sulfonyl radical 6 reacted with alkyne 117 to give the vinyl radical 119. Next, the radical intermediate 120 was formed via the intramolecular spirocyclization of the vinyl radical with an aryl ring. Finally, the oxidation of 120 produced the oxygenium intermediate 121, which would provide 3-sulfonyl azaspiro[4,5]trienone 118. In 2020, Wei group also reported a visible-light-induced protocol for the construction of sulfonylated benzofurans through oxidative cyclization of 1, 6-enynes and arylsulfinic acids, in which the C–S, C-C and C=O bonds was sequentially formed in one pot procedure [64].

|
Download:
|
| Scheme 35. Visible-light-induced difunctionalization of activated alkynes with sulfinic acids for the construction of 3-sulfonyl azaspiro[4,5]trienones. | |
{kind=link}
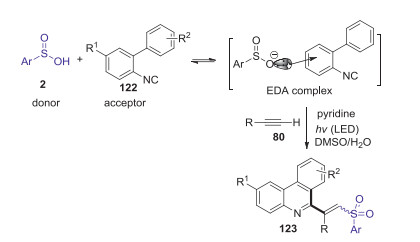
Phenanthridines have drawn considerable synthetic interest of chemists owing to their important applications in medicinal chemistry and materials science. In 2018, Wang and co-workers presented photochemical synthesis of C6 polyfunctionalized phenanthridines through three-component reaction of sulfinic acids, isocyanides, and alkynes [65]. This simple reaction provides good yields of the desired products with abroad substrate scope and excellent selectivity. The mechanism studies suggested that this reaction was induced by the photochemical activity of the novel electron donor-acceptor (EDA) complex, which was generated from the reaction of biaryl isocyanide and arylsulfinic acid in the presence of water and pyridine under mild conditions (Scheme 36).

|
Download:
|
| Scheme 36. Photochemical synthesis of C6 polyfunctionalized phenanthridines from three-component reaction of sulfinic acids, isocyanides, and alkynes. | |
{kind=link}
In 2018, a copper-catalyzed decarboxylative disulfonylation of alkynyl carboxylic acids with sulfinic acids was developed by Li group [66]. The reaction exhibits good stereoselectivity and favorable functional group tolerance, providing a straightforward and practical approach to access various (E)-1, 2-disulfonylethenes in good yields. A possible mechanism is showed in Scheme 37. Firstly, sulfonyl radical 6 was generated from sulfinic acid 2 in the presence of ammonium persulfate. On the other hand, the decarboxylation of alkynyl carboxylic acid with copper salt gave the alkynyl copper species 126. Subsequently, addition of the sulfonyl radical 6 to the alkynyl copper 126 produced radical 127, which further interacted with the sulfonyl radical 6 to afford intermediate 128. Finally, the protonation of intermediate 128 afforded the desired disulfonylethene.

|
Download:
|
| Scheme 37. Copper-catalyzed decarboxylative disulfonylation of alkynyl carboxylic acids with sulfinic acids. | |
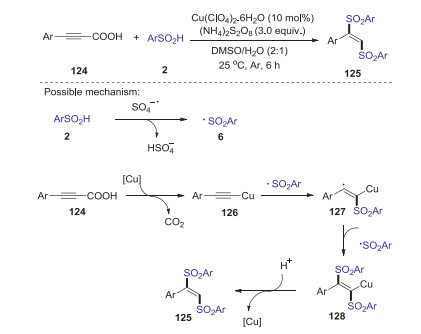{kind=link}
A facile AcOH/tert-butyl nitrite promoted oxidative intermolecular sulfonamination of alkynes with sulfinic acids to prepare sulfonyl pyrroles was reported by Yan group [67]. This tandem addition/cyclization reaction was conducted well by using of tert-butyl nitrite as the oxidant, in which various substituted sulfonyl pyrroles were obtained in moderate to good yields without of metal reagents. A proposed reaction pathway is showed in Scheme 38. Initially, sulfinic acids 2 reacted with TBN to generate the corresponding sulfonyl radical 6. Subsequently, the sulfonyl radical 6 added to the alkynyl moiety of 129 to give the vinyl radical intermediate 131. The intramolecular radical addition and cyclization intermediate 131 gave intermediate 132. Finally, 132 is oxidized by TBN to afford the desired product 130.

|
Download:
|
| Scheme 38. tert-Butyl nitrite promoted oxidative intermolecular sulfonamination of alkynes with sulfinic acids. | |
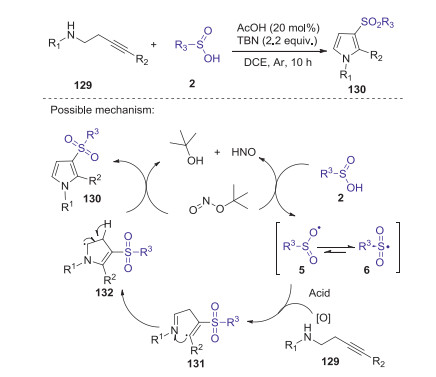{kind=link}
In 2018, Wang and Meng developed a convenient synthetic strategy for the synthesis of sulfonyl substituted furans through an O2-oxidative radical tandem cycloaddition of enynones with arylsulfinic acids [68]. Various arylsulfinic acids with either an electron-rich or -poor group on the aromatic rings could be efficiently converted to corresponding products in good yields with favorable functional group tolerance. Based on the control experiments, a possible reaction mechanism is proposed in Scheme 39. Firstly, sulfinic acid 2 was decomposed in water to generate sulfinyl anion 4, which was transformed into sulfonyl radical 6 under air. Subsequently, sulfonyl radical 6 attacked enynone 133 to yield an enolate radical 135, which underwent an intramolecular cyclization to afford an radical intermediate 136. Finally, the hydrogen abstraction of intermediate 136 from H2O or 2 afforded desired product 126.

|
Download:
|
| Scheme 39. tert-Butyl nitrite promoted oxidative intermolecular sulfonamination of alkynes with sulfinic acids. | |
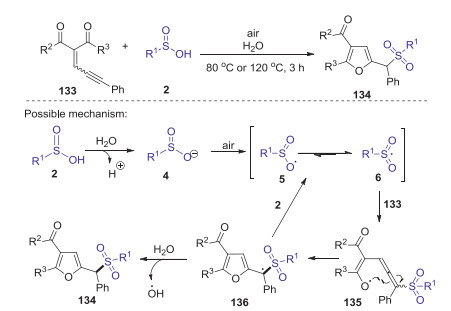{kind=link}
In 2019, Li's group reported an efficient pyridine mediated oxidative radical cyclization of N-propargyl anilines with sulfinic acids leading to 3-sulfonated quinolines under the visible light irradiation (380–385 nm) [69]. This reaction could be carried out under external photocatalyst-free conditions using air as an ideal oxidant, which provided a facile approach to 3-sulfonated quinoline derivatives with good yields, excellent functional group tolerance, and high regio-selectivity (Scheme 40).

|
Download:
|
| Scheme 40. Pyridine mediated oxidative radical cyclization of N-propargyl anilines with sulfinic acids leading to 3-sulfonated quinolines. | |
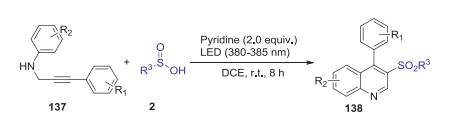{kind=link}
Very recently, Zhou reported a novel TBPB-initiated cascade cyclization of 3-arylethynyl-[1, 1′-biphenyl]−2-carbonitriles with sulfinic acids for the synthesis of 3-sulfonated cyclopenta[gh] phenanthridines under metal-free conditions [70]. This transformation was achieved under mild conditions through the tandem C–S/C–C/C–N bond formation in one pot procedure. A proposed reaction pathway is presented in Scheme 41. Initially, thermal decomposition of TBPB gave radicals Ⅰ and Ⅱ, which abstracted a hydrogen from the sulfinic acid to generate a sulfonyl radical 6. Subsequently, the addition of sulfonyl radical 6 to 139 afforded intermediate 141, which underwent rapid intra-molecular cyclization to produce the iminyl radical 142. Then, intramolecular addition of iminyl radical to the pendant aromatic ring formed radical intermediate 143. Next, the oxidation of 143 produced the corresponding cation 144, which was deprotonated to give the desired product 140 (path a). The other possible pathway to produce the desired product is that the radicals Ⅰ and Ⅱ abstracted a hydrogen atom from the intermediate 143 (path b).

|
Download:
|
| Scheme 41. TBPB-initiated cascade cyclization of 3-arylethynyl-[1, 1′-biphenyl]-2-carbonitriles with sulfinic acids. | |
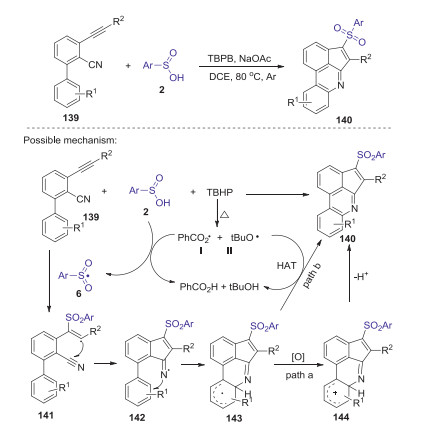{kind=link}
2.3. Sulfonylation of allenes
As a valuable framework containing two cumulative C-C double bonds, allenes have been widely employed in the synthesis of important biologically active compounds and natural products [71]. Lei and co-workers reported a highly regio- and stereoselective pyridine mediated oxysulfonylation of allenes with arylsulfinic acids and dioxygen [72]. Various 2-sulfonyl allylic alcohols were obtained in satisfactory yields under mild metal-free conditions with good functional group tolerance. Preliminary mechanistic studies indicated that a radical process might be involved in this reaction and hydroxyl oxygen atom of product came from dioxygen in air. The detailed reaction mechanism is presented in Scheme 42. Initially, sulfonyl anion 4 was generated from arylsulfinic acid in the presence of pyridine. Subsequently, the autoxidation of 4 by dioxygen afforded an oxygen-centered radical 5 resonating with sulfonyl radical 6. Then, the addition of sulfonyl radical 6 to allene gave the reactive allyl radical 147, which interacted with dioxygen to form intermediate 148. Next, the intermediate 148 underwent through the SET and PT process successively with 4 and pyridinium, giving peroxide 150. Finally, the expected product 146 was produced through the subsequent reduction process.

|
Download:
|
| Scheme 42. Oxysulfonylation of allenes with sulfinic acids and dioxygen. | |
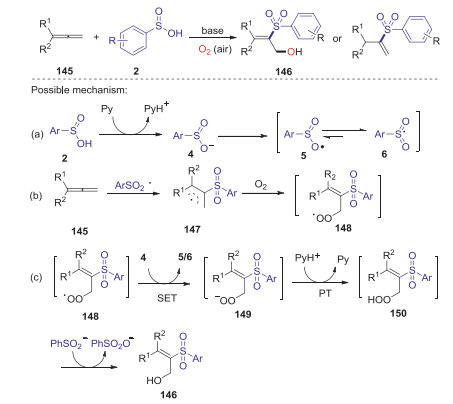{kind=link}
2.4. Sulfonylation of arenes
Functionalized quinolines are widely existed in various natural products, functional materials, and biologically active molecules. In 2019, He and co-workers reported a visible-light mediated organic dye-catalyzed method for the construction of 2-sulfonylquinoline via deoxygenative C2-sulfonylation of quinoline N-oxides with sulfinic acid [73]. This protocol employing air as the green oxidant and acetone/water as the solvent, provides a mild and efficient route to access 2-sulfonylquinolines in good to excellent yields. This reaction could also be carried out in a scaled-up manner allowing late-stage modification of biologically active compounds containing quinoline motifs.
A proposed reaction pathway is shown in Scheme 43. Firstly, sulfonyl radical 6 was formed from sulfinic acid 2 in the presence of Na2·Eosin Y and dioxygen under visible-light irradiation. Subsequently, quinoline N-oxide 151 reacted with the sulfonyl radical 6 to afford an intermediate 153, which was further trapped by sulfonyl free-radical 6 to afford the intermediate 154. Finally, intermediate 154 underwent dehydro-aromatization to give the expected 2-sulfonylquino-line 152 with the release of sulfonic acid.

|
Download:
|
| Scheme 43. Visible-light mediated synthesis of 2-sulfonylquinoline via deoxygenative C2-sulfonylation of quinoline N-oxides with sulfinic acid. | |
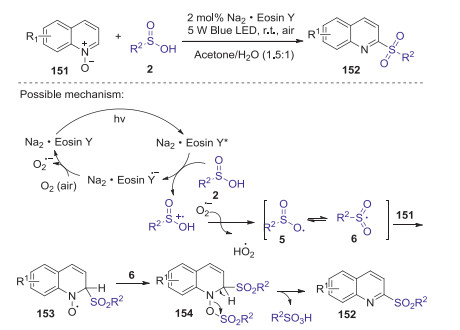{kind=link}
At the same time, Wang group also presented a visible-light-induced and Na2·Eosin Y catalyzed deoxygenative C2-sulfonylation of quinoline N-oxides with sulfinic acids (Scheme 44) [74]. This protocol shows a broad substrate scope and functional group tolerance, and desired products with various substituents could be obtained in moderate to good yields at room temperature by using of TBHP as the oxidant. Similar to the He's work, mechanistic studies suggested that the radical process was also involved in this transformation.

|
Download:
|
| Scheme 44. Visible-light-induced deoxygenative C2-sulfonylation of quinoline N-oxides with sulfinic acids in the presence of TBHP. | |
{kind=link}
2.5. Sulfonylation of alcohols
The direct utilization of easily accessible alcohols and sulfinic acids as precursors to construct organic sulfones is particularly attractive due to that generate water as the only byproduct. In 2018, Loh and Xie reported a regiospecific dehydrative cross-coupling reaction between sulfinic acids and propargyl alcohols to access propargylic sulfones under catalyst- and additive-free conditions [75]. A series of propargyl sulfones could be constructed in high yields with good regioselectivities from a wide range of alcohols and sulfinic acids. The mechanism is not yet understood in detail, and a possible pathway is proposed as shown in Scheme 45. Initially, intermediate 157 was formed through the hydrogen bond between the sulfinic acid and propargylic alcohol. Then, a γ-selective attack by an oxygen atom of the sulfinic acid gave intermediate 158 via a bridged-ring transition state (TS1). Subsequently, intermediate 158 rapidly produced racemic product 156 under the acidic conditions through favored sigmatropic rearrangement process.

|
Download:
|
| Scheme 45. Regiospecific dehydrative cross-coupling reaction between sulfinic acids and propargyl alcohols to access propargylic sulfones. | |
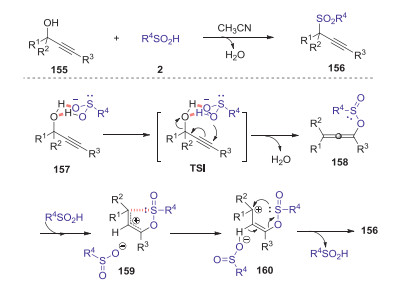{kind=link}
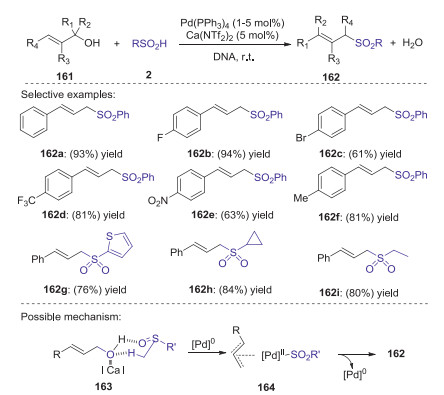
In 2020, Loh and Xie described Pd(PPh3)4 and Ca(NTf2)2 co-catalyzed strategy for the synthesis of allylic sulfones through the allylic sulfination with unactivated allylic alcohols with sulfinic acids [76]. In this procedure, the hydrogen bond interaction between sulfinic acids and allylic alcohols enabled a dehydrative cross-coupling process to give a variety of allylic sulfones in good to excellent yields under mild reaction conditions (Scheme 46). Remarkably, the reaction can be conducted on a gram scale, in which allylic sulfones could be isolated without chromatography. Preliminary studies indicated that the calcium salt was not indispensable for this process. Calcium salt might facilitate the formation of intermediate 163, which was followed by the insertion of palladium that gave the key intermediate 164. The eductive elimination of palladium from intermediate 164 afforded the desired 162.

|
Download:
|
| Scheme 46. Synthesis of allylic sulfones via the allylic sulfination with unactivated allylic alcohols with sulfinic acids. | |
{kind=link}
Very recently, Zhou group also reported a mild method for the dehydrative synthesis of allylic sulfones via Pd-catalyzed sulfonylation of allylic alcohols with sulfinic acids in aqueous media [77]. Various allylic sulfones would be efficiently obtained from non-derivatized allylic alcohols and sulfinic acids by simple use of the easily-available Pd(PPh3)4 as the catalyst. Mechanism studies suggested two possible reaction pathways might be involved in this transformation (Scheme 47). The path a (in common aprotic organic media) involves a substrate self-assisted activation of the allylic alcohol via a six-membered ring intermediate 165 that generated from sulfinic acid 2 and allylic alcohol 161. The path b (in aqueous media) involves an eight-membered ring intermediate 169 that formed from sulfinic acids, allylic alcohols, and water via hydrogen bonding interaction.

|
Download:
|
| Scheme 47. Pd-catalyzed sulfonylation of allylic alcohols with sulfinic acids in aqueous media. | |
{kind=link}
2.6. Others
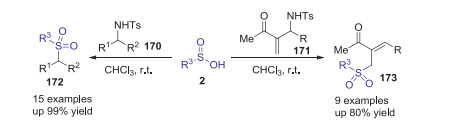
In 2009, Tian and co-workers reported a catalyst-free sulfonylation of N-benzylic and N-allylic sulfonamides with sulfinic acids via sp3 C-N bond cleavage at room temperature [78]. Through this strategy, various structurally diversified sulfones were prepared in moderate to excellent yields in the absence of any external catalysts and additives (Scheme 48). It should be noted that the reaction of sulfinic acids with N-(2-acyl)allylic sulfonamides offered a convenient approach to access trisubstituted allyl sulfones with exclusive Z selectivity.

|
Download:
|
| Scheme 48. Sulfonylation of N-benzylic and N-allylic sulfonamides with sulfinic acids via C(sp3) CN bond cleavage. | |
{kind=link}
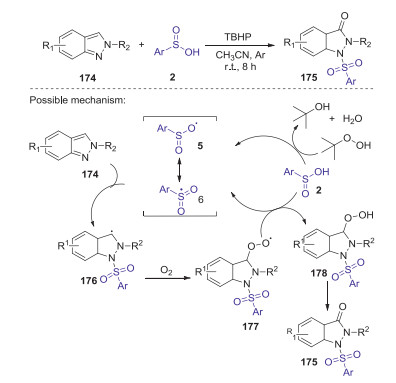
Sulfonamides have significant applications in pharmaceutical chemistry and industrial research. Recently, Hajra group reported a new and facile protocol for the construction of N-sulfonylated indazolones via oxo-sulfonylation of indazolone with sulfinic acid under ambient air (Scheme 49) [79]. This method provides a series of structurally diverse 1-sulfonylindazol-3(2H)-one derivatives with good yields by using of tert-butyl hydroperoxide as oxidant, which has the advantages of broad substrate suitability, mild conditions, and scalability. A proposed reaction pathway is presented in Scheme 49. Initially, sulfinic radical 6 was generated from sulfinic acid 2 in the presence of TBHP. Then, carbon intermediate 176 was produced via the addition of the sulfinic radical 6 to N-1 position of 2H-indazole 174. Subsequently, intermediate 176 at the C-3 position of 2H-indazole was oxidized by dioxygen to give intermediate 177, which abstracted a hydrogen radical from sulfinic acid to afford intermediate 178. Finally, the elimination of water from the intermediate 178 produced the desired product 175.

|
Download:
|
| Scheme 49. Oxo-sulfonylation of indazolone with sulfinic acid leading to N-sulfonylated indazolones. | |
{kind=link}
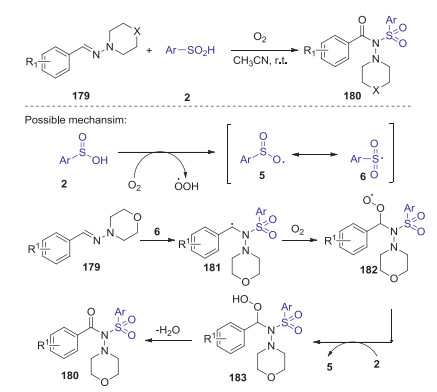
In 2020, Hajra and co-workers also reported a simple metal-free method for the synthesis of N-acylsulfonamides through oxo-sulfonylation of aldehyde-derived hydrazones with sulfinic acid [80]. A wide range of functionalized N-acylsulfonamides could be efficiently obtained through C-O and Np-S bond-forming reaction at room temperature, in which dioxygen was used as a green oxidant. A proposed reaction pathway is described in Scheme 50. Initially, sulfinic acid generated sulfonyl radical 6 in the presence of molecular oxygen. Next, the addition of sulfonyl radical 6 at the iminium nitrogen center of hydrazones 179 to give the intermediate 181. Subsequently, interaction of intermediate 181 with dioxygen afforded intermediate 182, which is converted to intermediate 183 via the hydrogen radical abstraction from sulfinic acid 2. Finally, the desired N-acylsulfonamides 180 was obtained through the elimination of water from 183.

|
Download:
|
| Scheme 50. Oxo-sulfonylation of aldehyde-derived hydrazones with sulfinic acid to access N-acylsulfonamides. | |
{kind=link}
3. Sulfinylation
Sulfoxide is an important and versatile building block that is widely existed in various valuable natural products and materials. In 2015, Wang reported a novel strategy for the synthesis of 3-arylsulfinylindoles via direct sulfenylation of indoles with arylsulfinic acids [81]. This reaction could be conducted under metal- and additive-free conditions to provide an attractive approach to a series of 3-arylsulfinylindoles with 50%−95% yields at ambient temperature in water. Preliminary mechanistic mechanism using electrospray ionization mass spectrometry indicated that an electrophilic substitution of the sulfinyl cation process was involved in this transformation (Scheme 51).

|
Download:
|
| Scheme 51. Sulfenylation of indoles with arylsulfinic acids leading to 3-arylsulfinylindoles. | |
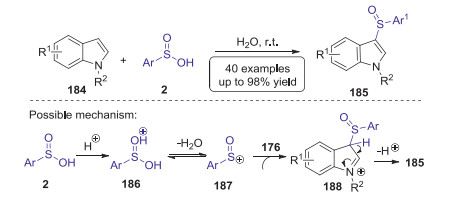{kind=link}
Later, Wang and Miao presented a mild BF3-promoted selective preparation of diarylsulfoxides and m-arylthiosulfones from arylsulfinic acids and arenes under mild reaction conditions [82]. When the reactions of sulfinic acids and arenes were carried out in CH2Cl2 at 30 ℃ under air, the corresponding diarylsulfoxides were selectively obtained in good yields with favorable functional group tolerance via an unusual sulfinyl cation (Scheme 52).

|
Download:
|
| Scheme 52. BF3-promoted C-S bond formation for selective synthesis of diaryl sulfoxides. | |
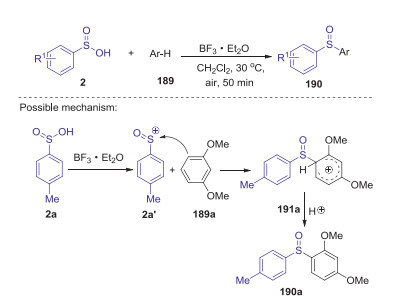{kind=link}
Interestingly, when the reactions of sulfinic acids and arenes were conducted in CH2Cl2 at 80 ℃ under N2 for 2.5 h, which enabled the generation of two different sulfur-containing groups at the aromatic rings with high regioselectivity and a range of structurally diverse m-arylthio sulfones were obtained in good yields. Mechanistic studies suggested m-arylthio sulfones were formed through the reaction of diaryl sulfoxides with sulfinyl cation by a sequence of redox process and electrophilic aromatic substitution reaction (Scheme 53).

|
Download:
|
| Scheme 53. BF3-promoted selective synthesis of m-arylthio sulfones from arylsulfinic acids and arenes. | |
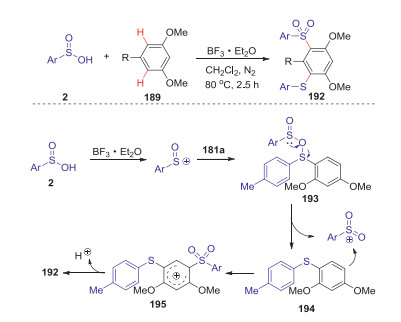{kind=link}
4. Sulfenylation
Sulfinic acids can also be employed as sulfenylating agents to construct various sulfides in synthetic chemistry. In 2015, Liu group developed an effective method to synthesize 3-arylthioindoles and 3-alkylthioindoles via regioselective sulfenylation of indoles with sulfinic acids [83]. A number of aryl- and alkyl-sulfinic acids as well as indoles bearing either electron-withdrawing or electron-donating groups on the indole rings are suitable substrates to give structurally diverse indole thioethers with good to excellent yields in the presence of TsOH (10 mol%) and n-Bu4NI (1.2 equiv.). The byproduct I2 played as an efficient catalyst to promote this sulfenylation reaction. Plausible mechanism is showed in Scheme 54. Initially, TsOH and n-Bu4NI promoted the reduction of sulfinic acid 2 to give the disulfide 198 with the release of I2. Then, disulfide 198 reacted with I2 produced the sulfenyl iodide 199, which interacted with indole 196 to give 3-sulfenylindole 197 and HI.

|
Download:
|
| Scheme 54. Sulfenylation of indoles with sulfinic acids to construct 3-thioindoles. | |
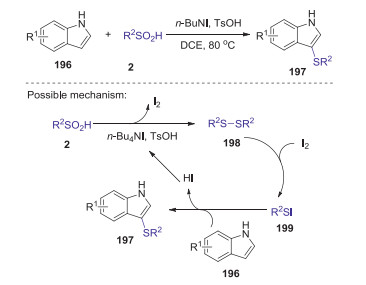{kind=link}
β-Alkoxy sulfides are highly valuable intermediates in synthetic and medicinal chemistry. In 2016, Yan and Lin reported a new method for the synthesis of β-alkoxy sulfides through a NaI/HBr-promoted three-component oxysulfenylation reaction of alkenes with alcohols and arylsulfinic acids [84]. This reaction could be conducted under transition-metal free conditions to give various β-alkoxysulfides in good yields. A plausible mechanism is showed in Scheme 55. Firstly, the reaction of NaI with HBr generated HI. Subsequently, sulfinic acid 2 was reduced by HI to form disulfide 198 and iodine. Then, disulfide 198 reacted with iodine to afford an electrophilic species Ar2SI 199, which added to alkene 1 giving a thiiranium ion 202. Finally, the interaction of intermediate 202 with ROH 200 afforded product 201 and HI.

|
Download:
|
| Scheme 55. NaI/HBr-promoted three-component oxysulfenylation reaction of alkenes with alcohols and arylsulfinic acids. | |
{kind=link}
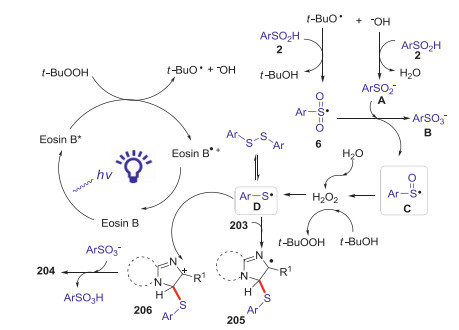
In 2017, Yang reported a visible-light-induced C-H sulfenylation of imidazoheterocycles with sulfinic acid for the synthesis of C-3 sulfenylated imidazoheterocycles under irradiation by a 3 W blue-light LED lamp [85]. This protocol using Eosin B as the cheap photocatalyst and arylsulfinic acids as odorless sulfur reagents, provides a novel approach toward the synthesis of heteroaryl sulfides. Under standard conditions, a range of sulfinic acids with either electron-withdrawing or electron-donating groups, were all efficiently converted to the corresponding C-3 sulfenylated imidazoheterocycles in good to excellent yields (Scheme 56). This transformation demonstrates a new model for C–S bond formation through a photoredox process.

|
Download:
|
| Scheme 56. Visible light-induced C-H sulfenylation of imidazoheterocycles with sulfinic acid. | |
{kind=link}
The detailed mechanism is demonstrated in Scheme 57. Firstly, the excited species Eosin B* was generated from the photocatalyst Eosin B by visible light irradiation. Then, the single electron transfer (SET) from Eosin B* to TBHP giving a hydroxyl anion and tert-butoxyl radical. The interaction of tert-butoxyl radical with arylsulfinic acid 2 provided the sulfonyl radical 6, which reacted with arylsulfinate A to give the sulfonic acid anion B and sulfinyl radical C. Subsequently, the reduction of C by t-BuOH or H2O afforded the thiyl radical D, which added to the imidazoheterocycle to form the carbon-centered radical 205. Then, the single electron transfer (SET) from 205 to Eosin B·+ gave the carbocation intermediate 206. Finally, the β-H of intermediate 206 was attacked by the sulfonic acid anion produced the desired product 204.

|
Download:
|
| Scheme 57. The possible mechanism for the visible-light-induced C-H sulfenylation of imidazoheterocycles with sulfinic acid. | |
{kind=link}
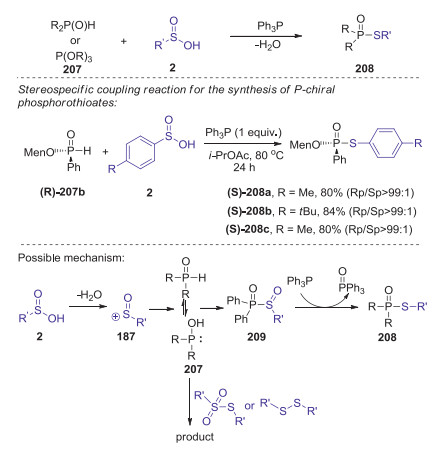
In 2017, Hong presented a novel reduction coupling strategy for the preparation of phosphorothioates from H-phosphoryl compounds and sulfinic acids under metal- and oxidant-free conditions [86]. This S-P(O) bond formation reaction was realized under metal-, oxidant-, and halogen-free conditions by the addition of PPh3 as a reductant. This method is compatible with many substituents on a number of sulfinic acids including halogens and heterocyclic moieties. Moreover, optically active P-chiral phosphorothioates could be selectively synthesized via stereo specific coupling reactions. A plausible mechanism is showed in Scheme 58. Initially, a sulfinyl cation was formed in situ by the dehydration of sulfinic acid. Then, the tautomerization of H-phosphoryl compounds P(Ⅳ) to P(Ⅲ), which attacked a sulfinyl cation to give intermediate 209. Other possible pathway involving sulfonothioate or disulfide reactive species might also be involved in this transformation. Finally, the reduction of intermediate 209 by triphenyl phosphine afforded the desired product 208.

|
Download:
|
| Scheme 58. Synthesis of phosphorothioates via reduction coupling of H-phosphoryl compounds with sulfinic acids. | |
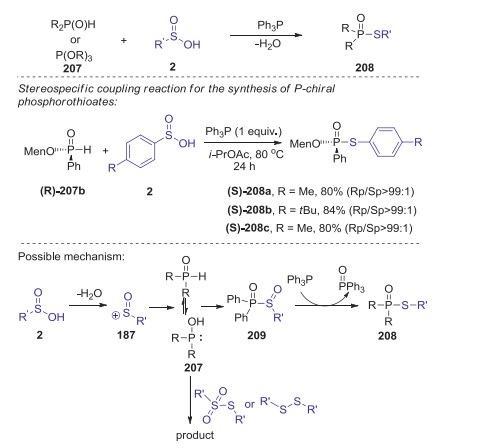{kind=link}
5. Conclusion
In recent years, sulfinic acids as odorless, readily available and versatile sulfur-reagents have been increasingly utilized in a variety of synthetic transformations. This research area is emerging one of the most attractive and appealing protocol for the construction of diverse sulfur-containing compounds with high atom economy. In this review, we mainly describe three reaction patterns of sulfinic acids and their corresponding reaction mechanism. In this regard, most synthetic strategies are focused on sulfonylation process via the formation of either sulfonyl radical or anions reactive species. The sulfinylations or sulfenylations involving sulfinyl cation/radical or disulfide have also been described.
Despite the notable advances have been achieved in recent years, there are still some challenges waiting to be explored in the future. For example, most sulfonylation reactions are limited to the arylsulfinic acids. Extremely scarce examples have been realized involving the utilization of alkylsulfinic acids, which makes the application of more practical in the synthesis of sulfur-containing molecules. Furthermore, the development of milder and safer strategies such as photocatalysis or electrocatalysis will be highly desirable to avoid the use of potentially dangerous oxidants. Additionally, direct C(sp3)-H functionalization and asymmetric reaction modes has not been fully disclosed, which is expected to be developed in the near future. It is strongly believed that further investigation will eventually make the utilization of sulfinic acids become one of the most valuable and practical protocols for the synthesis of organosulfur compounds owing to their important applications in synthetic chemistry, pharmaceutical and materials.
Declaration of competing interestWe declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
AcknowledgmentsThis work was supported by the Youth Innovation Technology Project of Higher School in Shandong Province (No. 2019KJC021), Qinghai Science and Technology Achievement Transformation Project (No. 2019-SF-122), and Qinghai Key Laboratory of Tibetan Medicine Research (No. 2021-ZJ-Y03).
| [1] |
I.P. Beletskaya, V.P. Ananikov, Chem. Rev. 111 (2011) 1596-1636. DOI:10.1021/cr100347k |
| [2] |
C.P. Ashcroft, P. Hellier, A. Pettman, S. Watkinson, Org. Process Res. Dev. 15 (2011) 98-103. DOI:10.1021/op100251q |
| [3] |
M. Feng, B. Tang, S.H. Liang, X. Jiang, Curr. Top. Med. Chem. 16 (2016) 1200-1216. DOI:10.2174/1568026615666150915111741 |
| [4] |
H.J.M. Gijsen, M.A.J. De Cleyn, M. Surkyn, et al., Bioorg. Med. Chem. Lett. 22 (2012) 547-552. DOI:10.1016/j.bmcl.2011.10.091 |
| [5] |
A. EI-Awa, M.N. Noshi, X.M. du Jourdin, P.L. Fuchs, Chem. Rev. 109 (2009) 2315-2349. DOI:10.1021/cr800309r |
| [6] |
Z. Gan, G. Li, X. Yang, et al., Sci. China. Chem. 63 (2020) 1652-1658. DOI:10.1007/s11426-020-9811-6 |
| [7] |
S. Zhao, K. Chen, L. Zhang, W. Yang, D. Huang, Adv. Synth. Catal. 362 (2020) 3516-3541. DOI:10.1002/adsc.202000466 |
| [8] |
S. Ye, G. Qiu, J. Wu, Chem. Commun. 55 (2019) 1013-1019. DOI:10.1039/C8CC09250H |
| [9] |
C. Shen, P. Zhang, Q. Sun, et al., Chem. Soc. Rev. 44 (2015) 291-314. DOI:10.1039/C4CS00239C |
| [10] |
F. Dénès, C.H. Schiesser, P. Renaud, Chem. Soc. Rev. 42 (2013) 7900-7942. DOI:10.1039/c3cs60143a |
| [11] |
Y. Wu, Y.W. Lin, W.M. He, Chin. Chem. Lett. 31 (2020) 2999-3000. DOI:10.1016/j.cclet.2020.09.005 |
| [12] |
J. Aziz, A. Hamze, Org. Biomol. Chem. 18 (2020) 9136-9159. DOI:10.1039/D0OB01718C |
| [13] |
L. Tang, K. Du, B. Yu, L. He, Chin. Chem. Lett. 31 (2020) 2991-2992. DOI:10.1016/j.cclet.2020.03.030 |
| [14] |
G. Qiu, K. Zhou, J. Wu, Chem. Commun. 54 (2018) 12561-12569. DOI:10.1039/C8CC07434H |
| [15] |
S. Ghosh, S. Samanta, A.K. Ghosh, S. Neogi, A. Hajra, Adv. Synth. Catal. 352 (2020) 4552-4578. |
| [16] |
S. Peng, Y.X. Song, J.Y. He, et al., Chin. Chem. Lett. 30 (2019) 2287-2290. DOI:10.1016/j.cclet.2019.08.002 |
| [17] |
J. Zhu, W. Yang, X Wang, L. Wu, Adv. Synth. Catal. 360 (2018) 386-400. DOI:10.1002/adsc.201701194 |
| [18] |
Q. Liu, L. Wang, H. Yue, et al., Green Chem 21 (2019) 1609-1613. DOI:10.1039/C9GC00222G |
| [19] |
G. Li, H. Xie, J. Chen, Y. Guo, G.J. Deng, Green Chem. 19 (2017) 4043-4047. DOI:10.1039/C7GC01932G |
| [20] |
Z. Xu, H. Huang, H. Chen, G.J. Deng, Org. Chem. Front. 6 (2019) 3060-3064. DOI:10.1039/C9QO00592G |
| [21] |
Y. Li, J.P. Wan, Chin. J. Org. Chem. 40 (2020) 3889-3894. DOI:10.6023/cjoc202005026 |
| [22] |
Y. Guo, G. Wang, L. Wei, J.P. Wan, J. Org. Chem. 84 (2019) 2984-2990. DOI:10.1021/acs.joc.8b02897 |
| [23] |
Q. Liu, Y. Lv, R. Liu, et al., Chin. Chem. Lett. 32 (2021) 136-139. DOI:10.1016/j.cclet.2020.11.059 |
| [24] |
W.H. Bao, Z. Wang, X. Tang, et al., Chin. Chem. Lett. 30 (2019) 2259-2262. DOI:10.1016/j.cclet.2019.06.052 |
| [25] |
J. Aziz, S. Messaoudi, M. Alami, A. Hamze, Org. Biomol. Chem. 12 (2014) 9743-9759. DOI:10.1039/C4OB01727G |
| [26] |
Y.Q. Jiang, J. Li, Z.W. Feng, et al., Adv. Synth. Catal. 362 (2020) 2609-2614. DOI:10.1002/adsc.202000233 |
| [27] |
Z.W. Feng, J. Li, Y.Q. Jiang, et al., New J. Chem. 44 (2020) 14786-14790. DOI:10.1039/D0NJ03386C |
| [28] |
K. Sun, X.L. Chen, Y.L. Zhang, et al., Chem. Commun. 55 (2019) 12615-12618. DOI:10.1039/C9CC06924K |
| [29] |
M.D. McReynolds, J.M. Dougherty, P.R. Hanson, Chem. Rev. 104 (2004) 2239-2258. DOI:10.1021/cr020109k |
| [30] |
Q.A. Acton, Sulfones-Advances in Research and Application, Scholarly Editions, Atlanta, 2013.
|
| [31] |
N.S. Simpkins, Sulfones in Organic Synthesis. New York: Pergamon Press, 1993.
|
| [32] |
Q. Lu, J. Zhang, F. Wei, et al., Angew. Chem. Int. Ed. 52 (2013) 7156-7159. DOI:10.1002/anie.201301634 |
| [33] |
C.V. Galliford, K.A. Scheidt, Angew. Chem. Int. Ed. 46 (2007) 8748-8758. DOI:10.1002/anie.200701342 |
| [34] |
T. Shen, Y. Yuan, S. Song, N. Jiao, Chem. Commun. 50 (2014) 4115-4118. DOI:10.1039/c4cc00401a |
| [35] |
W. Wei, J. Wen, D. Yang, et al., Green Chem. 16 (2014) 2988-2991. DOI:10.1039/C4GC00231H |
| [36] |
W. Wei, J. Wen, D. Yang, et al., Org. Biomol. Chem. 12 (2014) 7678-7681. DOI:10.1039/C4OB01369G |
| [37] |
Q. Lu, J. Zhang, P. Peng, et al., Chem. Sci. 6 (2015) 4851-4854. DOI:10.1039/C5SC00807G |
| [38] |
Z. Yuan, H.Y. Wang, X. Mu, et al., J. Am. Chem. Soc. 137 (2015) 2468-2471. DOI:10.1021/ja5131676 |
| [39] |
G. Zhang, L. Zhang, H. Yi, et al., Chem. Commun. 52 (2016) 10407-10410. DOI:10.1039/C6CC04109D |
| [40] |
D. Yang, B. Huang, W. Wei, et al., Green Chem. 18 (2016) 5630-5634. DOI:10.1039/C6GC01403H |
| [41] |
J.D. Scottand, R.M. Williams, Chem. Rev. 102 (2002) 1669-1730. DOI:10.1021/cr010212u |
| [42] |
F. Chen, N.N. Zhou, J.L. Zhan, B. Han, W. Yu, Org. Chem. Front. 4 (2017) 135-139. DOI:10.1039/C6QO00535G |
| [43] |
D. Xia, Y. Li, T. Miao, P. Li, L. Wang, Chem. Commun. 52 (2016) 11559-11562. DOI:10.1039/C6CC04983D |
| [44] |
P. Qian, Y. Deng, H. Mei, et al., Org. Lett. 19 (2017) 4798-4801. DOI:10.1021/acs.orglett.7b02163 |
| [45] |
M.H. Huang, Y.L. Zhu, W.J. Hao, et al., Adv. Synth. Catal. 361 (2019) 5534-5539. DOI:10.1002/adsc.201901212 |
| [46] |
Y.L. Zhu, C.F. Zhu, P. Zhou, et al., J. Org. Chem. 83 (2018) 9641-9653. DOI:10.1021/acs.joc.8b00994 |
| [47] |
Z.J. Shen, Y.N. Wu, C.L. He, et al., Chem. Commun. 54 (2018) 445-448. DOI:10.1039/C7CC08516H |
| [48] |
Z. Wu, R. Ren, C. Zhu, Angew. Chem. Int. Ed. 55 (2016) 10821-10824. DOI:10.1002/anie.201605130 |
| [49] |
M.W. Zheng, X. Yuan, Y.S. Cui, et al., Org. Lett. 20 (2018) 7784-7789. DOI:10.1021/acs.orglett.8b03191 |
| [50] |
H.S. Dutta, A. Ahmad, A.A. Khan, et al., Adv. Synth. Catal. 361 (2019) 5534-5539. DOI:10.1002/adsc.201901212 |
| [51] |
Q. Liu, Y. Mei, L. Wang, Y. Ma, P. Li, Adv. Synth. Catal. 362 (2020) 5669-5680. DOI:10.1002/adsc.202000846 |
| [52] |
G.H. Lia, Q.Q. Han, Y.Y. Sun, et al., Chin. Chem. Lett. 31 (2020) 3255-3258. DOI:10.1016/j.cclet.2020.03.007 |
| [53] |
Q. Zhang, D. Dong, W. Zi, J. Am. Chem. Soc. 142 (2020) 15860-15869. DOI:10.1021/jacs.0c05976 |
| [54] |
Q. Lu, J. Zhang, G. Zhao, et al., J. Am. Chem. Soc. 135 (2013) 1148-1154. DOI:10.1021/ja311497e |
| [55] |
W. Wei, J. Li, D. Yang, et al., Org. Biomol. Chem. 12 (2014) 1861-1864. DOI:10.1039/C3OB42522C |
| [56] |
Y. Xi, B. Dong, E.J. McClain, et al., Angew. Chem. Int. Ed. 53 (2014) 4657-4661. DOI:10.1002/anie.201310142 |
| [57] |
W. Yang, S. Yang, P. Lia, L. Wang, Chem. Commun. 51 (2015) 7520-7523. DOI:10.1039/C5CC00878F |
| [58] |
F. Chen, Q. Meng, S.Q. Han, B. Han, Org. Lett. 18 (2016) 3330-3333. DOI:10.1021/acs.orglett.6b01427 |
| [59] |
N. Zhou, Z. Yan, H. Zhang, Z. Wu, C. Zhu, J. Org. Chem. 81 (2016) 12181-12188. DOI:10.1021/acs.joc.6b01847 |
| [60] |
C. Wu, P. Yang, Z. Fu, et al., J. Org. Chem. 81 (2016) 10664-10671. DOI:10.1021/acs.joc.6b01549 |
| [61] |
J. Wen, W. Shi, F. Zhang, et al., Org. Lett. 19 (2017) 3131-3134. DOI:10.1021/acs.orglett.7b01256 |
| [62] |
J. Xu, X. Yu, J. Yan, Q. Song, Org. Lett. 19 (2017) 6292-6295. DOI:10.1021/acs.orglett.7b02971 |
| [63] |
W. Wei, H. Cui, D. Yang, et al., Green Chem. 19 (2017) 5608-5613. DOI:10.1039/C7GC02330H |
| [64] |
L. Wang, M. Zhang, Y. Zhang, et al., Chin. Chem. Lett. 31 (2020) 67-70. DOI:10.1016/j.cclet.2019.05.041 |
| [65] |
Y. Li, T. Miao, P. Li, L. Wang, Org. Lett. 20 (2018) 1735-1739. DOI:10.1021/acs.orglett.8b00171 |
| [66] |
H. Fu, J.Q. Shang, T. Yang, et al., Org. Lett. 20 (2018) 489-492. DOI:10.1021/acs.orglett.7b03922 |
| [67] |
Z. Qi, Y. Jiang, Y. Wang, R. Yan, J. Org. Chem. 83 (2018) 8636-8644. DOI:10.1021/acs.joc.8b00741 |
| [68] |
Y. Ren, L.G. Meng, T. Peng, L. Wang, Org. Lett. 20 (2018) 4430-4433. DOI:10.1021/acs.orglett.8b01714 |
| [69] |
Y. Zhang, W. Chen, X. Jia, L. Wang, P. Li, Chem. Commun. 55 (2019) 2785-2788. DOI:10.1039/C8CC10235J |
| [70] |
N. Zhou, M. Wu, M. Zhang, X. Zhou, W. Zhou, Org. Biomol. Chem. 18 (2020) 1733-1737. DOI:10.1039/D0OB00119H |
| [71] |
S. Ma, Acc. Chem. Res. 42 (2009) 1679-1688. DOI:10.1021/ar900153r |
| [72] |
Z. Huang, Q. Lu, Y. Liu, et al., Org. Lett. 18 (2016) 3940-3943. DOI:10.1021/acs.orglett.6b01575 |
| [73] |
L.Y. Xie, T.G. Fang, J.X. Tan, et al., Green Chem. 21 (2019) 3858-3863. DOI:10.1039/C9GC01175G |
| [74] |
D.Q. Dong, L.X. Li, G.H. Li, et al., Chin. J. Catal. 40 (2019) 1494-1498. DOI:10.1016/S1872-2067(19)63420-0 |
| [75] |
Y. Liu, P. Xie, Z. Sun, et al., Org. Lett. 20 (2018) 5353-5535. DOI:10.1021/acs.orglett.8b02188 |
| [76] |
P. Xie, Z. Sun, S. Li, et al., Org. Lett. 22 (2020) 4893-4897. DOI:10.1021/acs.orglett.0c01747 |
| [77] |
J. Yu, X. Chang, R. Ma, Eur. J. Org. Chem. (2020) 7238-7242. |
| [78] |
C.R. Liu, M.B. Li, D.J. Cheng, C.F. Yang, S.K. Tian, Org. Lett. 11 (2009) 2543-2545. DOI:10.1021/ol900788r |
| [79] |
P. Ghosh, S. Mondal, A. Hajra, Org. Lett. 22 (2020) 1086-1090. DOI:10.1021/acs.orglett.9b04617 |
| [80] |
S. Mondal, A.K. Ghosh, A. Hajra, Org. Lett. 22 (2020) 2771-2775. DOI:10.1021/acs.orglett.0c00759 |
| [81] |
T. Miao, P. Li, Y. Zhang, L. Wang, Org. Lett. 17 (2015) 832-835. DOI:10.1021/ol503659t |
| [82] |
W. Shi, T. Miao, Y. Li, P. Li, L. Wang, Org. Lett. 20 (2018) 4416-4420. DOI:10.1021/acs.orglett.8b01681 |
| [83] |
C.R. Liu, L.H. Ding, Org. Biomol. Chem. 13 (2015) 2251-2254. DOI:10.1039/C4OB02575J |
| [84] |
D. Wang, R. Zhang, W. Ning, Z. Yan, S. Lin, Org. Biomol. Chem. 14 (2016) 5136-5140. DOI:10.1039/C6OB00646A |
| [85] |
P. Sun, D. Yang, W. Wei, et al., Green Chem. 19 (2017) 4785-4791. DOI:10.1039/C7GC01891F |
| [86] |
Y. Moon, Y. Moon, H. Choi, S. Hong, Green Chem. 19 (2017) 1005-1013. DOI:10.1039/C6GC03285K |