 2022, Vol. 33
2022, Vol. 33 


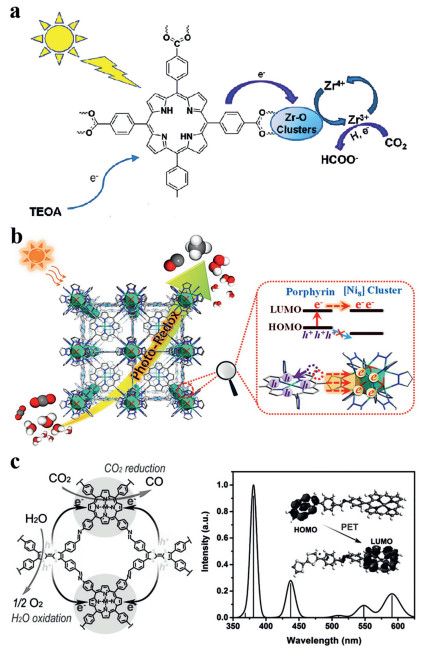
Approximately 120, 000 TW of solar energy can reach the surface of the earth each year, which is 8000 times the total global energy demand [1]. Solar energy is dispersive and unstable, so how to better transform and store solar energy is the key to the use of solar energy for solving the energy crisis. Inspired by the photosynthesis of green plants in nature, the use of photocatalytic technology to directly convert solar energy into chemical energy in chemical substances is essential for the sustainable development of mankind [2-7]. Generally speaking, a complete photocatalytic reaction usually includes the following steps. One is the capture of photons and then the conversion of photons to electrons, which are generally completed by semiconductors or organic chromophores with appropriate energy band structures [8, 9]. The spectral response range and photoelectric conversion capacity of the light capture unit determine the upper limit of the photocatalytic activity. The second is the exciton separation and the transport of photogenerated carriers, that is, a part of excitons are separated by spontaneous diffusion or the action of an electric field, then the photogenerated electrons and holes generated by the separation diffuse to the vicinity of the catalyst surface through the catalyst body. In order to transmit as many photogenerated carriers as possible to the catalyst surface, long carrier lifetime, short diffusion distance, and weak diffusion resistance are usually required. The third is the surface reaction, that is, the reaction substrate is adsorbed on the surface of the activated catalyst [10-17]. The activated substrate accepts the photogenerated carriers on the surface of the catalyst, then being converted at the active sites, so the content and properties of active sites have a great influence on the activity and selectivity of photocatalysis [18-20].
The existing photocatalysts can be roughly divided into heterogeneous catalysts (TiO2 [21-26], CdS [27, 28], C3N4 [29-32], COFs [33-35], MOFs [36-39], etc.) and homogeneous catalysts formed by metal ions and organic ligands (such as FeEs4 [40], CuPS@CoTMPyP [41], ZnP-phen=Re [42]). Porphyrin compounds are currently the most widely studied tetrapyrrole compounds, with a large molar absorption coefficient, high fluorescence quantum yield, large Stokes shift, and excellent thermal stability [43-49]. In the photosynthetic reaction of plants and bacteria, the chromophores capable of converting solar energy into chemical energy are mainly based on porphyrins [50]. In addition, the energy band structures and electronic properties of porphyrin derivatives can be adjusted by the surrounding substituents and metal coordination centers. Homogeneous photocatalysts often have ideal activity and selectivity due to their molecular-level dispersed active sites and excellent mass transfer performances [51-53]. Because the catalyst can be dispersed at the molecular level in the system, its catalytic performance is easily affected by the surrounding chemical environment, and its stability is relatively poor [54-56].
Compared with homogeneous catalysts, heterogeneous catalysts can overcome the disadvantage that homogeneous catalysts are difficult to recover [57]. However, photocatalytic reactions usually only occur at the surface of heterogeneous photocatalysts, thus their active centers are often not well utilized. In addition, the heterogeneous catalysts generally exist in bulk, which is not conducive to light capture and substance transmission. Moreover, the chemical microenvironment of each part of the heterogeneous catalyst is different, which will greatly affect the selectivity of the photocatalytic reaction.
In order to improve light absorption capacity and the separation efficiency of photogenerated carriers, as well as the stability of the heterogeneous catalyst, some small organic molecules are introduced in their photocatalytic systems as the photosensitizers or active sites. Wang et al. synthesized a TiO2 nanosheets/tetra (4-carboxyphenyl)porphyrin (TiO2 NSs/TCPP) hybrid system for CO2 photoreduction via a simple self-assembly approach. The photocatalytic performance of the TiO2 NSs/11.5%TCPP was 37 times higher than that of the pristine TiO2 NSs for the reduction of CO2 to CO [26]. Lu et al. synthesized Zinc 5, 10, 15, 20-meso-tetra(4-hydrazidephenyl)porphyrin (ZnTHPP)/CdS nanosheets (CdS NSs) by a hydrothermal method. Due to the presence of the functional group of acylhydrazine in ZnTHPP, the rate constant for photogenerated holes was about 1.7 times higher than the pure CdS NSs, and exhibited excellent photostability (15 h) and efficient photocatalytic activity for pure water splitting (6.4 times than CdS NSs) [27]. g-C3N4 has good chemical stability, but the serious carrier recombination limits further improvement of its photocatalytic performance. Combining porphyrin derivatives with carbon nitride is a feasible method. Ye et al. firstly reported Co-porphyrin/low-molecular-weight g-C3N4 photocatalyst through the covalent bonding for the photoreduction of CO2 to CO. Due to effective electron transfer and capture of Co active sites, and the affinity of Co-porphyrin and CO2, the photocatalytic efficiency of the hybrid (17 µmol g−1 h−1) was 13 times higher than that of bulk g-C3N4 [30].
In recent years, covalent organic frameworks and metal organic frameworks have received extensive attention due to their large specific surface areas, adjustable pores, and band structures. However, these framework materials are still faced with difficulty in separating and transporting photogenerated carriers. In order to solve these problems, inorganic materials such as metal clusters [58-72], metallic compounds [73-76], organic molecule [77-83]. were coupled with the framework materials to build heterojunctions or increase active sites. Do et al. combined a porphyrin-based metal organic framework (Al/PMOF) with amine-functionalized graphene (NH2-rGO) as the photocatalyst for the photoreduction of CO2 to HCOO‒. The conversion rate of NH2-rGO(5 wt%)/Al-PMOF was 75.87% higher than that of Al-PMOF, and its photocatalytic activity was as high as 685.5 µmol g−1 h−1 with almost 100% selectivity [38].
Regardless of whether porphyrin is used as a photosensitizer or a catalytic center, the current porphyrin-based photocatalysts often have the problem of poor interfacial bonding between porphyrin and other components, which is very unfavorable for the separation and transportation of the photogenerated carriers. Additionally, because artificial photocatalytic systems often lack a non-photochemical quenching (NPQ) process similar to nature, the excessive photon flux will destroy the catalyst stability [84].
In this review, we reviewed the problems encountered in the research of porphyrin-based photocatalysts as well as solutions developed in recent years, and clarified the inner principles of these methods, and emphasized the importance of molecular design for heterogeneous catalysts (Fig. 1). Firstly, the porphyrin end group modification and metal center regulation from the molecular level were summarized. Secondly, interface engineering plays an important role in high-performance heterogeneous porphyrin-based photocatalysts. Thus, the interface engineering of porphyrin derivatives and inorganic photocatalysts including molecular bridge, metal bridge, and non-contact sensitization was also summarized. Thirdly, we proposed the strategy for spatial confinement of porphyrin derivatives in the organic frameworks. Finally, the remaining challenges and future prospects of porphyrin-based heterogeneous photocatalysts were discussed.

|
Download:
|
| Fig. 1. Design of heterogeneous porphyrin-based molecular photocatalyst. | |
Porphyrin-based homogeneous photocatalysis has received extensive attention due to its higher activity and selectivity. Relevant researchers have constructed a variety of porphyrin-based homogeneous photocatalysts with excellent performances through molecular design, but these homogeneous photocatalysts were often difficult to recover together with poor stability [54, 57]. Therefore, the heterogeneity of homogeneous catalysts through molecular self-assembly or composite is one of the current development trends for photocatalysts. In this case, if the existing molecular design theory of porphyrin-based homogeneous photocatalysts could be introduced into the study of porphyrin-based heterogeneous photocatalysts, the photocatalytic performances of porphyrin-based heterogeneous photocatalysts will be significantly improved (Fig. 2). Therefore, here we will introduce the important influence of the molecular design of porphyrin on the photocatalytic reaction from terminal groups modification and metal centers regulation of porphyrin.

|
Download:
|
| Fig. 2. Schematic diagram of molecular design of porphyrin. | |
It is well known that the energy band structure and molecular polarity of porphyrin derivatives can be regulated by adjusting the terminal groups [23, 85-90]. In addition, due to the special electron distribution of porphyrins, the meso-substituted porphyrins are generally more favorable for electron transfer than the β-substituted porphyrins [91, 92]. On the basis of meso-substituted porphyrins, we proposed three methods: (1) Increasing the electron absorption capacity of the terminal groups; (2) expanding the conjugated structure of porphyrin molecules; (3) increasing the affinity of the non-anchor groups to the reaction substrates.
When n-type semiconductors are in close contact with p-type semiconductors, a robust microscopic electric field will be formed at the interfaces [93-95]. This persistent microscopic electric field is similar to the dipole moment in organic molecules, so strong dipole moments often correspond to strong intramolecular charge transfer driving forces. In other words, if the porphyrin ring was modified with a strong electron-absorbing terminal group, a dipole moment appeared between the porphyrin ring and the terminal group, which would facilitate the electron transport within the porphyrin ring.
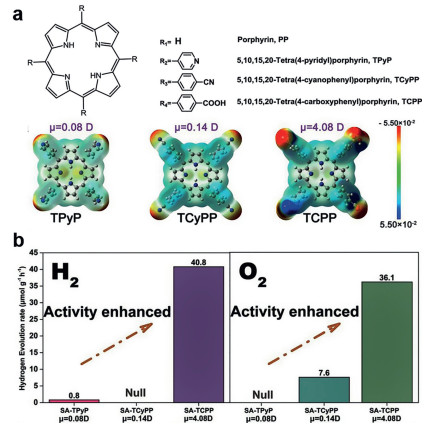
Wang et al. synthesized three self-assembled porphyrin photocatalysts using three porphyrin derivatives with different polar end groups, and studied the relationship between the photocatalytic performance and molecular dipole moment (Fig. 3a). The hydrogen production and oxygen production performance of TCPP with the maximum dipole moment were 40.8 µmol g−1 h−1 and 36.1 µmol g−1 h−1 respectively (Fig. 3b). Then Lu et al. also used the DFT method to calculate and simulate the electron density distribution of three porphyrin derivatives containing different end groups (Fig. 4a), and the results showed that the polar groups stretched the molecular structure of the porphyrin [96]. Under the action of the carboxyl group, the energy band structure of the molecule did not change drastically although the electrons moved from the center of the porphyrin ring to the benzene ring [86]. It can be seen that the internal dipole moment generated by the strong polar groups is very important for the internal carrier separation and transfer of porphyrin molecules during the process of photocatalytic total hydrolysis. In addition, strong polar groups also increase the hydrophilicity of self-assembled porphyrins, which is also an important factor for affecting the activity of catalysts.

|
Download:
|
| Fig. 3. (a) Molecular structures and Dipole moment of different porphyrin molecules. (b) Photocatalytic hydrogen evolution and oxygen evolution without cocatalyst. Reproduced with permission [86]. Copyright 2018, Wiley-VCH. | |

|
Download:
|
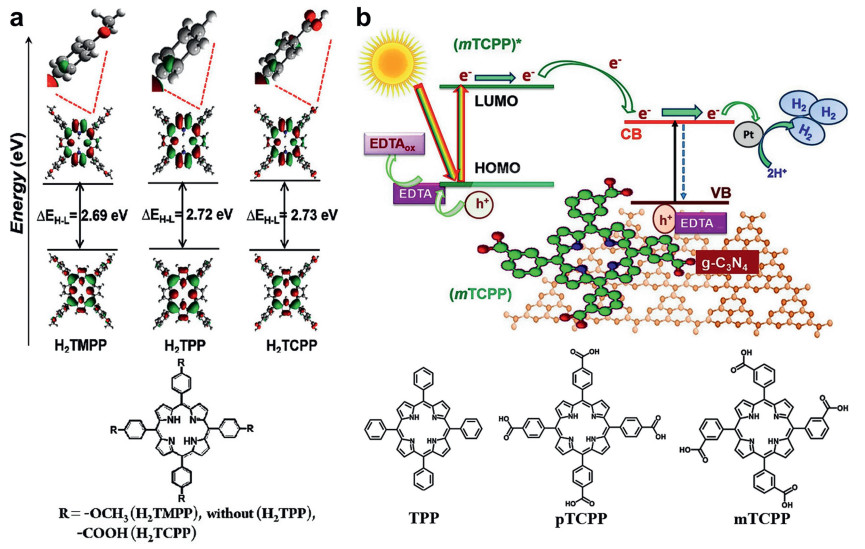
| Fig. 4. (a) Molecular structure and frontier molecular orbital profiles of H2TMPP, H2TPP, H2TCPP, all calculations performed at the DFT/TDDFT/B3LYP/6–311G(d)/LANL2DZ/level of theory. Reproduced with permission [96]. Copyright 2019 American Chemical Society. (b) Molecular structures of the semiconductor CN and of the porphyrins TPP, pTCPP, and mTCPP. Reproduced with permission [97]. Copyright 2017, Elsevier B.V. | |
Faria et al. used an impregnation method to combine TPP, mTCPP, and pTCPP (Fig. 4b) with carbon nitride by the non-covalent way to investigate the effects of carboxyl groups and their substitution sites on photocatalytic activities. Under the same conditions, the order of the photocatalytic activities of the three hybrid photocatalysts was: 17mTCPP (202 µmol) > 17pTCPP (191 µmol) > 17TPP (140 µmol) [97].
The reasons for the difference being that the larger molecular dipole moment is favorable for the separation and transfer of carriers in the porphyrin ring, and the terminal group reduces the formation of porphyrin aggregates and promotes the effective transfer of carriers between porphyrin and carbon nitride. In addition, the photocatalytic performance of mTCPP was higher than that of pTCPP due to its similar π-conjugated structure like carbon nitride, resulting in stronger binding with carbon nitride and higher carrier separation efficiency.
To sum up, firstly, improving the electron absorption ability of the end group can make the molecular structure be elongated, the electron concentration in the center of the porphyrin ring decreases, and the electron concentration near the end group increases. Secondly, the elongated molecular structure makes the LUMO energy level shift to the end group, which will effectively enhance the electron transfer tendency between the LUMO energy level and the support surface. At the same time, the decrease of electron concentration in the porphyrin ring can also effectively increase the interaction between porphyrin and electron-rich support, which is conducive to the carrier transport between them. Finally, the increase of molecular polarity also enhances the affinity between photocatalyst and water molecules, which is particularly important for the photocatalytic reaction using water as substrate. For porphyrin-based photocatalysts anchored by a single terminal group, the remaining meso-substitutions can provide an opportunity to promote energy/carrier transport within porphyrins. We can not only prevent the excessive aggregation of porphyrins by modifying these sites, but also regulate the frontier orbitals of porphyrins.
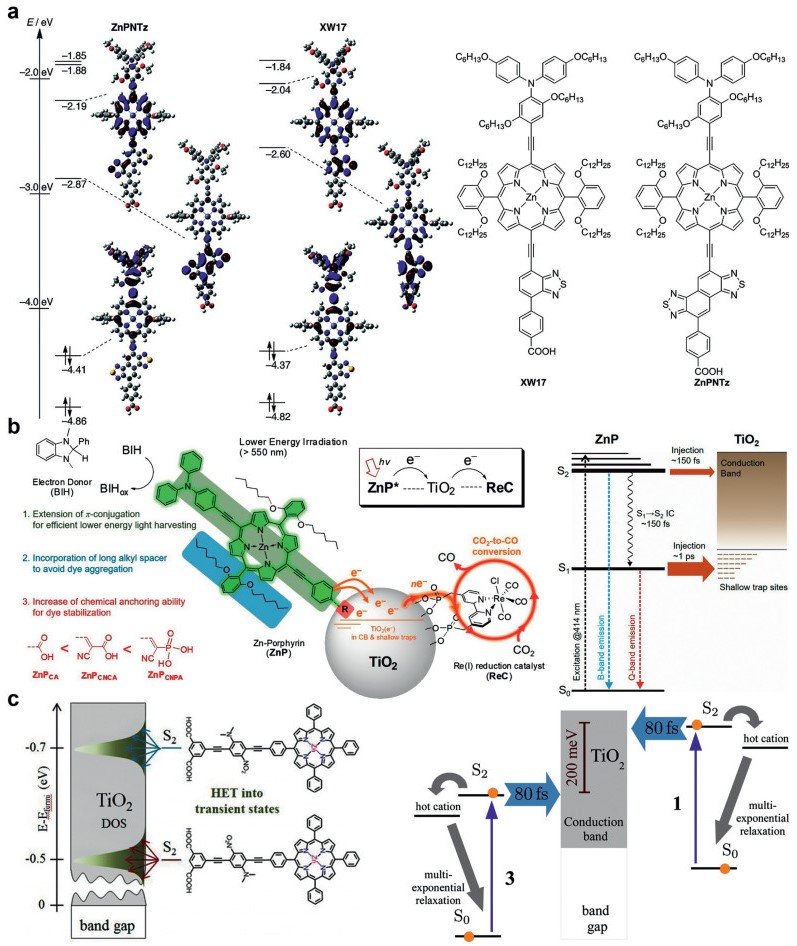
Imahori et al. reported two porphyrin sensitizers (Fig. 5a). The porphyrin ZnPNTz showed excellent absorption in the near-infrared (NIR) region as well as a large Stokes shift due to the naphtho[1, 2-c: 5, 6-c']bis[1, 2, 5]thiadiazole moiety. The orbital distributions of the HOMOs were delocalized on the electron-donating triarylamine moiety and porphyrin core owing to effective π expansion via the ethynyl linkage. In contrast, the orbital distributions of the LUMOs were mainly localized on the electron-accepting NTz of ZnPNTz and BT of XW17 [98]. This asymmetric frontier orbital distribution will facilitate the transfer of electrons on LUMOs to the substrate. The time-dependent DFT (TD-DFT) calculations showed that the lowest-energy excitation mainly possessed a HOMO→LUMO transition, corresponding to the Q-band. The excitation energy of ZnPNTz (1.36 eV) was less than that of XW17 (1.50 eV), which well agreed with the redshift of the Q-band.

|
Download:
|
| Fig. 5. (a) Molecular structure and energy diagrams and selected Kohn-Sham orbitals of model D-π-A porphyrins on the optimized structures. Reproduced with permission [98]. Copyright 2018, Wiley-VCH. (b) Sequential energy/electron transfer process in a dye-sensitized hybrid system from lower energy porphyrin photosensitizers (ZnPs) to TiO2 (molecular immobilizer and electron mediator) and Re(I) reduction catalyst (ReC). Reproduced with permission [84]. Copyright 2018, American Chemical Society. (c) Jablonski diagram for 1 and 3 on the TiO2 film including HET. The energy axis is not to scale. Reproduced with permission [99]. Copyright 2016, American Chemical Society. | |
Recently, Kang et al. synthesized a series of Zn-porphyrin molecules supported on the surface of TiO2/ReC. The different anchoring groups (COOH, CNCOOH, CNPO3H2) were utilized to test their binding capability with TiO2. All porphyrin molecular hybrids exhibited good photocatalytic activities under low energy irradiation (> 550 nm). The porphyrin with a phosphoric acid group (ZnPCA) has the strongest binding force with TiO2, thus possessing the highest photocatalytic stability. Porphyrin (ZnPCNPA) has the highest photocatalytic efficiency due to the special sp2 hybridization of the carboxyl group and its excellent electron-withdrawing ability (TONRe was over 1028). The overall photophysical reaction pathways are summarized in Fig. 5b [84].
More importantly, a more efficient carrier injection from porphyrin to support could be achieved when we associate this extended porphyrin structure with molecular polarity. Pal et al. used zinc porphyrin (LG5) with a similar structure to compound with TiO2, showing apparent quantum yield (AQY) of 7.43% and photocatalytic activity of 4196 µmol g−1 h−1 with a turnover number of 8392 [22]. Gundlach et al. investigated the heterogeneous electron transfer (HET) between photoexcited molecules and colloidal TiO2 for several zinc porphyrin chromophores anchored onto TiO2 via reorientation of the dipole moment. These unique dye molecules were studied by femtosecond transient absorption spectroscopy in solution and on a TiO2 colloidal film. On the film, heterogeneous electron transfer was found to occur in 80 fs, much faster than all intramolecular pathways (Fig. 5c). It can be interpreted that HET resulted in the distribution of transition states that differ from regular surface states or bridge-mediated states [99].
As we all know, the affinity of the catalyst to the reaction substrate will significantly affect its photocatalytic activity and selectivity [100-102]. Zhang et al. reported an asymmetrically structured porphyrin (ZnPy)/TiO2 photocatalyst, and catalytic efficiency of 24.17 µmol g−1 h−1 was obtained for the reduction of CO2 to CH4 in the gas-phase environment. However, due to the lack of sacrificial agent in the gaseous environment, it will produce some problems such as the photogenerated carrier's self-recombination of ZnPy (marked as ④ in Fig. 6a), the injected electrons in TiO2's CB recombination with the oxidized ZnPy (marked as ⑤ in Fig. 6a) and oxidation of CO2 reduction products on ZnPy (marked as ⑥ in Fig. 6a) [25].

|
Download:
|
| Fig. 6. (a) The possible mechanism of CO2 photoreduction to CO/CH4 generation over the ZnPy-sensitized TiO2. Reproduced with permission [25]. Copyright 2015, Royal Society of Chemistry. (b) Experimental values of MO energy levels of ZnMT3PyP and ZnMTPP, VB/CB levels of g-C3N4, and redox potentials of sacrificial reagents. (c) Photocatalytic process of ZnMT3PyP/g-C3N4 and ZnMTPP/g-C3N4. (b, c) Reproduced with permission [103]. Copyright 2017, American Chemical Society. (d) Photocatalytic process of the molecular orbital energy levels of the ZnPy derivatives. Reproduced with permission [104]. Copyright 2020, Royal Society of Chemistry. | |
These harmful processes clearly show the important role of sacrificial agents in the process of photocatalysis, so how to enhance the interaction between sacrificial agents and catalysts for improving the activity and stability of photocatalyst has become a problem that relevant researchers must consider. Li et al. compared the photocatalytic performances of two porphyrins (ZnMTPP and ZnMT3PyP) hybrid systems with carbon nitride, and the ECB and EVB of two composites showed not obviously different.
In addition, ethylene diamine tetraacetic acid (EDTA), triethanolamine (TEOA), and ascorbic acid (AA) were added into the photocatalytic system as sacrificial agents to compare the effects of different sacrificial agents on the photocatalytic reaction (Fig. 6b). The results show that ZnMT3PyP/C3N4 has higher photocatalytic efficiency and stability than ZnMT3PyP/C3N4 when AA is used as a sacrificial agent. The reason is that AA can form hydrogen bonds with the pyridine nitrogen (Fig. 6c) at the edge of ZnMT3PyP to accelerate the carrier transport, but cannot form hydrogen bonds with the phenyl group at the edge of ZnMTPP [103].
Recently, Peng et al. synthesized three asymmetric zinc porphyrin derivatives (ZnPy-1, ZnPy-5, and ZnPy-6), and compared the performances of hybrid photocatalysts formed by the three porphyrin derivatives with carbon nitride (Fig. 6d). Although the energy band positions of three porphyrin derivatives were very similar, the photocatalytic activities of ZnPy-5 and ZnPy-6 were significantly higher than that of ZnPy-1. The combination of hole scavenger and catalyst oxidation center is beneficial for the enhanced catalyst activity. In addition, the photocatalytic activity of ZnPy-6 was also higher than that of ZnPy-5, and it exhibited good cycle stability, meaning that the combination of hole scavenger and catalyst oxidation center is affected by steric hindrance [104].
In short, the enhancement of the electron absorption ability of the terminal group can effectively stretch the structure of porphyrin molecules, and further enhance the carrier transport between porphyrin and support (or reaction substrate). Secondly, through the expansion and asymmetric design of porphyrin conjugated structure, not only wider spectral response can be obtained, but also the directional transmission of photogenerated carriers can be facilitated. Finally, we briefly discuss the important role of introducing the molecular polarity into the expansion of porphyrin conjugated structure.
2.2. Regulation of coordination metal centersThe four nitrogen atoms in the porphyrin ring act as strong Lewis bases that can coordinate with more than 50 kinds of metal ions [105, 106]. Two types of metalloporphyrins can be formed based on the size of coordinating metal cations. When the metal ion radius is 55–80 pm, the metal ions are coordinated in the plane of porphyrin rings; However, when the metal ion radius is higher than 80–90 pm, the out-of-plane or sitting-atop (SAT) metalloporphyrins can be formed [107, 108].
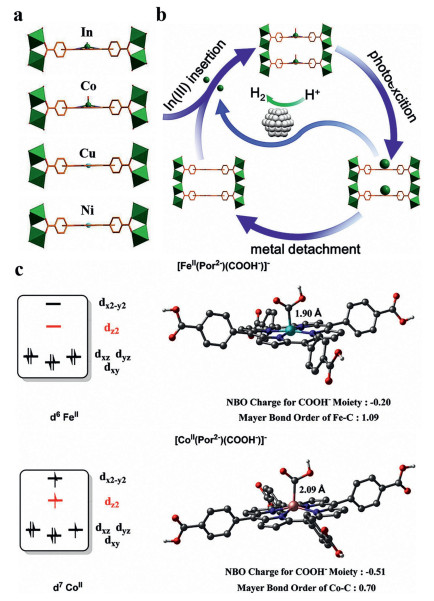
Jiang et al. synthesized a porphyrin organic framework with In3+ coordination. In the center of porphyrin, In(OH)3 acted as a precursor to control the reaction rate. The photocatalytic efficiency of the USTC-8 (100% In) was around 18–37 times higher than that of USTC-8 (M) (M = Co, Cu, Ni), and 3.4 times for USTC-8 (20% In). At the same time, USTC-8 (100% In) also showed excellent photocatalytic cycle stability. The mechanism of the photocatalytic system is shown as follows: at first, In3+ and tetrakis (4-carboxyphenyl) porphyrin (TCPP) coordinate to form sitting-atop (SAT) metalloporphyrin (Fig. 7a). Then, the exciting porphyrin molecules transfer electrons to In3+, causing the reduced In3+ to leave the porphyrin molecules. Finally, the electrons of the reduced free In3+ transfer to the Pt catalyst for promoting hydrogen evolution reaction, at the same time, In3+ can be regained and coordinated with the porphyrin ring (Fig. 7b) [36].

|
Download:
|
| Fig. 7. (a) View of the partial structure of USTC-8(In), USTC-8(Co), USTC-8(Cu), and USTC-8(Ni), highlighting the metal location in metalloporphyrin. (b) Proposed detachment-insertion mechanism of USTC-8(In) during the photocatalytic recycle. (a, b) Reproduced with permission [36]. Copyright 2018, American Chemical Society. (c) Influence of the electron configurations of the metal centers on the metal-carbon interactions in [FeⅡ(Por2−)(COOH−)]− and [CoⅡ(Por2−)(COOH−)]−. Reproduced with permission [109]. Copyright 2020, American Chemical Society. | |
When porphyrins coordinate with metals, the metal ions will gain electrons from the pyrrole rings, making it easier to allow electrons to flow in a non-π system. More importantly, different metal ions have different electronic structures, which results in different redox potentials for metallic porphyrins containing different metal centers and the same porphyrin ligand [110-112].
Therefore, we can coordinate the transition metal with the pyridine nitrogen, and use the effective electron transfer process from the ligand to the metal center to make the metal center the reaction center [96, 113-116]. Because there are many ways to combine carbon dioxide molecules with active centers, so we can adjust the activity and selectivity of the photocatalytic reaction by controlling the type and valence of the metal center.
Lu et al. reported three different metal-organic frameworks using In(NO3)3 and metalloporphyrins with carboxyl groups (MTCPP; M = Fe, Co, In) in solvothermal conditions. Photocatalytic tests showed that the photocatalytic selectivities of the three metal-organic frameworks were almost at the same level, but the photocatalytic activity of In-FeTCPP-MOF was obviously higher than that of In-CoTCPP-MOF and In-InTCPP-MOF, and the maximum photocatalytic activity of In-FeTCPP-MOF could reach 3469 µmol/g for 24 h. Time-resolved PL spectroscopy and a sequence of photocurrent responses showed that the Fe metal centers exhibited a stronger quenching ability than that of the Co centers, meaning that the photoelectric separation efficiency of Fe sites was higher than that of Co sites. In addition, the EIS curve showed that the charge transfer resistance of Fe sites was also significantly lower than that of two other metal sites.
DFT calculations were proceeded using Fe and Co porphyrins at various spin states as the initial models, thus obtaining a deep insight into the different activities of In-Fe TCPP-MOF and its cobalt analogue. In the [CoⅡ(Por2‒)(COOH‒)]‒ with a distorted square-based pyramidal geometry, the CoⅡ 3d7 center possesses a half-filled dz orbital that weakens the coordination of the COOH‒ moiety from the z-direction. As for the singlet [FeⅡ(Por2‒)(COOH‒)]‒, its empty dz orbital makes it easier to accept the electron pair from the COOH– donor (Fig. 7c), as indicated by its larger Mayer bond order for the metal-carbon interaction (1.09 vs. 0.70). As a result, the negative charge of the COOH– moiety can be efficiently lowered, thus stabilizing the intermediate [109].
Because different metallic porphyrins have different properties, thus providing an opportunity to have synergistic effects between various metalloporphyrins. Peng et al. synthesized a series of porphyrin-based conjugated polymers (Zn-ZnDETPP, Co-CoDETPP and Zn-CoDETPP). Zn-CoDETPP showed the highest hydrogen evolution activity (43 µmol/h). The ultraviolet photoelectron spectra (UPS) and EIS polts showed that Zn-CoDETPP had the maximum negative conduction band maximum(CBM) top (ECB) value and the minimum impedance semicircle than others, exhibiting the improved electron transfer dynamics [117].
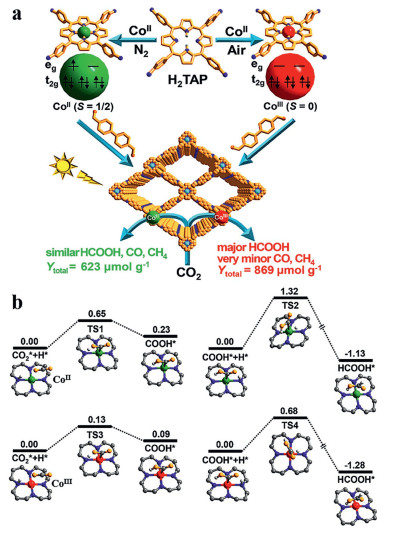
Moreover, the design of metalloporphyrin photocatalyst should be considered not only the appropriate metal type but also the valence state of the metal site. Recently, Jiang et al. reported two kinds of metal porphyrins with different oxidation states using Co(CH3COO)2·4H2O and CoCl2·6H2O as the precursors in nitrogen and air atmosphere respectively, thus obtaining two covalent organic frameworks (COF-367-CoⅢ, COF-367-CoⅡ) (Fig. 8a).

|
Download:
|
| Fig. 8. (a) Rational fabrication of COF-367-Co featuring different spin states of Co ions toward photocatalytic CO2 reduction. (b) Calculated potential energy profile of CO2 reduction reaction to HCOOH catalyzed by COF-367-CoⅡ and COF-367-CoⅢ. Reproduced with permission [118]. Copyright 2020, American Chemical Society. | |
DFT calculations showed that different oxidation states of Co corresponded to different electron spin states. Compared to COF-367-CoⅡ, COF-367-CoⅢ (CoⅢ, S = 0) had a lower energy barrier for the formation of HCOOH while a higher energy barrier for further conversion to CO and CH4 (Fig. 8b), thus leading to higher activity and significantly higher selectivity (producing HCOOH) than COF-367-CoⅡ (CoⅡ, S = 1/2) [118].
In conclusion, the properties of metalloporphyrins are not only affected by the types of metal centers but also modulated by the valence states of metal centers. First of all, different metals often have different radii and electronic structures, which is related to the binding mode of porphyrins and metal ions, leading to different charge transfer mechanisms between metal and ligand. Secondly, there is a synergy between different metal centers, and the use of mixed metal centers may achieve unexpected results. Finally, the different kinds and valence states of metals also lead to different spin states and distributions of electrons in the metal center, which will affect the evolution process of intermediates, and then affect the activity and selectivity of the photocatalytic reaction.
3. Interface engineering of porphyrin inorganic photocatalystSince Honda et al. first discovered the phenomenon of photolysis of titanium dioxide single crystals in 1972, inorganic photocatalysts have received great attention [119]. Although many inorganic materials such as TiO2 [21-23], MoS2 [120-122], and C3N4 [31, 97] have been applied to photocatalytic energy conversion, the activities and selectivities of these unmodified photocatalysts are still not satisfied (Table 1). Recently, porphyrins modified inorganic semiconductors have received more attention due to their excellent light absorption ability and adjustable redox potential. However, the matter of interface binding should be faced due to their inherent incompatibility of inorganic materials and organic molecules. In this part, strategies for improving the catalytic performances of porphyrin-based inorganic photocatalysts, such as molecular bridge, metal bridge, noncontact sensitization are mainly highlighted (Fig. 9). From the point of view of porphyrin molecular design, we will emphasize its important role in solving the problem of carrier transport at the interface of porphyrin inorganic photocatalyst.
|
|
Table 1 Summary of catalytic properties of porphyrin-based inorganic photocatalysts reported in recent years. |


|
Download:
|
| Fig. 9. Schematic diagram of the porphyrin-inorganic semiconductor. | |
In order to solve the interface incompatibility between inorganic materials and organic materials, researchers have introduced small organic molecules with flexible structures into inorganic-organic hybrid photocatalysts. Since the average distance of photocarriers in organic materials is about 20 nm, small organic molecules can be used as electronic buffers to reduce the back electron transfer due to the accumulation of photocarriers at the interface [86].
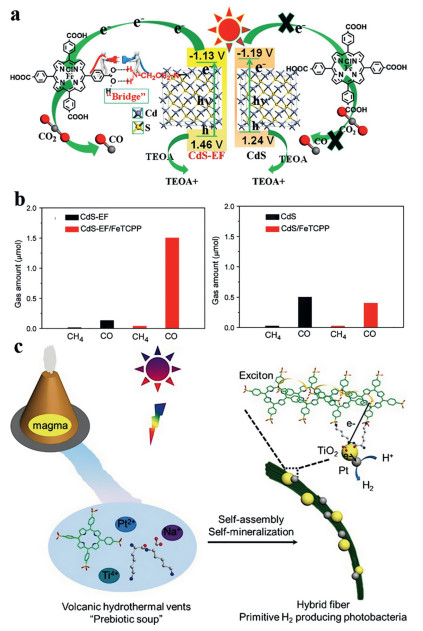
He et al. reported an example of Fe-porphyrin anchored onto CdS with/without functionalized ethylenediamine. After the functionalization, the bandgap of the hybrid increased from 2.43 eV to 2.59 eV, and the ECB changed from -1.19 to -1.13 V (Fig. 10a), which would weaken both the light absorption capacity and the driving force for the carrier migration. However, the photocatalytic efficiency of the former was much higher than that of the latter (Fig. 10b), because the carboxyl groups of the amino groups and porphyrin in ethylenediamine can form hydrogen bonds, thus promoting the carrier transport. In addition, in-situ synthesis of ethylenediamine functionalized CdS, nitrogen atoms can be well-coordinated with Cd to enhance the carrier transfer [123].

|
Download:
|
| Fig. 10. (a) Schematic diagram of CO2 photoreduction over CdS-EF/FeTCPP and CdS/FeTCPP hybrid catalysts. (b) CO2 photoreduction results over CdS-EF and CdS-EF/FeTCPP, and CdS and CdS/FeTCPP photocatalysts under visible-light illumination for 4 h. Reproduced with permission [123]. Copyright 2018, Elsevier B.V. (c) Illustration of the molecular evolution of porphyrins and peptides. A model of a primitive photosystem in a volcanic hydrothermal "prebiotic soup". Reproduced with permission [124]. Copyright 2016, Wiley-VCH. | |
Moreover, in-situ synthesis of organic small molecules/inorganic photocatalysts can also be realized through mineralization and electrostatic interaction. Recently, Yan et al. reported a bionic catalyst with a light funnel structure via self-assembly and biomineralization method (Fig. 10c). As TiO2 NPs do not directly bind to TPPS (tetrakis(4-sulfonatophenyl)porphine), the back electron transfer will be weakened [124]. TiO2 was synthesized through the electrostatic interaction and mineralization between TiBALDH (titanium(Ⅳ) bis(ammonium lactato)dihydroxide) and the peptide (l-Lys-l-Lys, KK), leading to strong interface binding between KK and TiO2. At the same time, the amino group of KK and the sulfonic group of porphyrin can form electrostatic interaction or hydrogen bond, promoting carrier migration. In addition, this hybrid photocatalyst also shows a synergistic effect between the three components and excellent cycle stability.
In conclusion, the molecular bridge can enhance the interface bonding between the inorganic part and the organic part of the catalyst through flexible end groups, thus improving the processability of the material [125-130]. Besides, the kinetics of charge transfer between the electron donors and acceptors can also be controlled through the structural design of the molecular chain [127, 131-134].
3.2. Construction of the metal bridgeIt is worth mentioning that there is a ligand-to-metal center transfer mechanism in metalloporphyrins. The shallow potential well-formed by the metal center can capture photogenerated electrons well. Therefore, the porphyrin can be anchored at the surface of the inorganic photocatalyst via the coordination between nitrogen atoms in the center of the porphyrin ring and the metal atoms at the surface of the semiconductor. The synergy between the two channels can optimize the photocatalytic activity and stability simultaneously [24, 135].
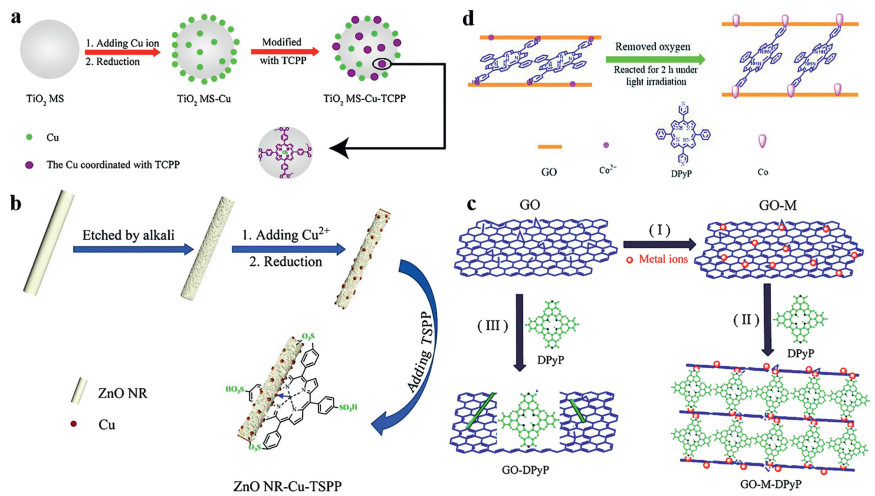
Li et al. used copper particles as an electron transport bridge to connect porphyrin to TiO2 (Fig. 11a), the photocatalytic activity of the porphyrin was 6 times higher than that of the original TiO2. The synergistic effect of central coordination and edge hydrogen bond improved the electron transfer between TiO2 and porphyrin [136].

|
Download:
|
| Fig. 11. (a) Schematic illustration for preparing the TiO2 MS-Cu-TCPP hybrid. Reproduced with permission [136]. Copyright 2016, Wiley-VCH. (b) Suggested assembly scheme of the ZnO NR-Cu-TSPP nanocomposites. Reproduced with permission [137]. Copyright 2020, Elsevier B.V. (c) Suggested formation schemes for the GO-DPyP and the GO-M-DPyP composites. Reproduced with permission [146]. Copyright 2016, Elsevier B.V. (d) The formation scheme of the GO-Co-DPyP nanohybrid. Reproduced with permission [147]. Copyright 2018, Royal Society of Chemistry. | |
To investigate the effects of the surrounding anchoring groups on the coordination of the central metal atoms in the porphyrin ring, Li et al. prepared porphyrins connected with ZnO using a copper bridge (Fig. 11b) and compared the photocatalytic properties of several porphyrins with different substituents. The results showed that the photocatalytic efficiency of sulfonic acid substituted porphyrins was 5.2 times higher than that of non-substituted porphyrins and 3.3 times higher than that of carboxyl substituents. The sulfonyl group has a stronger electron absorption capacity than the hydroxyl group or carboxyl group, so it is more favorable for carrier transport [137]. This indicates that the terminal groups of porphyrin still play an important role in the central metal coordination strategy of the porphyrin ring, at the same time, this synergistic effect can improve the activity and stability.
The strategy of constructing the metal bridge in the center of the porphyrin ring has also been applied to other inorganic catalysts such as NiO [135], Cu2O [24], and other inorganic photocatalyst systems. However, this strategy still fails to solve the problem of porphyrin aggregation when porphyrin was bonded with graphene or carbon nitride through the π-π interaction [138-144]. The obvious self-aggregation of porphyrins will seriously affect the stability and activity of the composite catalyst [145].
To solve this problem, it is better to build a metal bridge between the terminal group of the porphyrin and the support. Li et al. used Cr3+ to connect 5, 15-diphenyl-10, 20-di(4-pyridyl)porphyrin (DPyP) with graphene through electrostatic interaction and coordination (Fig. 11c). In the absence of metal ions on the surface, porphyrins mainly formed short rod-shaped J-aggregates. It is worth noting that a column supporting structure with an angle of less than 90° between the layers of graphene was formed after the addition of metal ions, which effectively decreased the occurrence of aggregation and charge transfer resistance. The GO-Cr3+-DPyP (928 µmol/g) had higher hydrogen evolution efficiency than GO (137 µmol/g) and GO-DPyP (686 µmol/g) for 8 h. Additionally, other metal ions (K+, Ca2+, Zn2+, Cu2+, and Co2+) also had positive impacts on the photocurrent response of the GO-DPyP hybrid systems [146].
In addition to using common metal ions as interface connectors, rare earth elements are also widely used in photocatalysis due to their unique electronic structures [148-150]. Li et al. introduced Ln3+ into 5, 15-diphenyl-10, 20-di(4-pyridyl) porphyrin (DPDPP)/GO, the obtained hybrid system of porphyrin with a column supporting angle of 38.3° It showed that there was an indirect Z-type heterojunction between porphyrin and GO. In addition, it was found that Sm3+ together with many rare earth metal ions (Gd3+, Ce4+, Eu3+, La3+, Nd3+, Er3+) had a similar influence on the photo-response of the hybrid [151].
It is reported that metal atoms usually have better photocatalytic efficiency than metal ions in photocatalysis. Li et al. compared the photocatalytic properties of the interface linker between 5, 15-diphenyl-10, 20-di(4-pyridyl)porphyrin (DPyP), and GO using Co2+ and Co atom respectively (Fig. 11d). There was only a small amount of Co2+ residue after the Co2+ was reduced by in-situ photoreduction, and the evolved hydrogen using the GO-Co-DPyP was 1093 µmol/g (irradiation for 2 h), which was about 2 times higher than that using the GO-Co2+-DPyP catalytic system [147]. Subsequent reports had also shown that this strategy of using atoms instead of ions as interface connectors could be used to construct metal bridges not only by the edges of porphyrin rings but also by the coordination between the ring centers and metal atoms [152, 153].
In conclusion, adding metal atoms or ions between porphyrin derivatives and supports can significantly reduce the interface resistance of porphyrins and supports, which has an electron relay effect. Secondly, the photocatalytic performance of the hybrid photocatalyst can also be controlled by the kind or valence of metal. Finally, for the porphyrin-based photocatalysts anchored by porphyrin ring, terminal group, and support, the type of terminal group also has an important influence on the photocatalytic performance.
3.3. Non-contact sensitizationIn recent years, related researchers have synthesized a series of porphyrin derivatives with long excited-state lifetimes through the structural design of porphyrin molecules [154-168], which provides a new idea for solving the interface problem of porphyrin and an inorganic compound.
Mourzina et al. synthesized a series of Sn porphyrin derivatives and supported them on the surface of TiO2 (Fig. 12a). Pre-adsorption of Sn(Ⅳ) meso-tetra(4-pyridyl)porphyrin dichloride (SnTPyP) onto TiO2 is a favorable factor for efficient hydrogen evolution in acidic media, while the maximum hydrogen yield was obtained for the SnTPyP sensitized TiO2/Pt at pH 9.0, indicating that the adsorption of SnTPyP onto the TiO2 surface was not essential for hydrogen evolution (Fig. 12b). More importantly, the photocatalytic activity of the SnTPyP/TiO2/Pt hybrid photocatalytic system was not completely lost in the environment with pH 3‒13 [23].

|
Download:
|
| Fig. 12. (a) Schematic diagram of SnP/TiO2 photocatalysis process. (b) Schematics of different pathways for hydrogen production. Reproduced with permission [23]. Copyright 2016, American Chemical Society. (c) Schematic illustration of the electron transfer dynamics occurring on a) SnP and b) Ru(dcbpy)3 sensitized TiO2 particle; The initial (1 h) H2 production rate in Ru(dcbpy)3/Pt-TiO2, SnP/Pt-sol and SnP/Pt-TiO2 suspensions as a function of pH. Reproduced with permission [169]. Copyright 2010, Royal Society of Chemistry. | |
In order to explain the difference between the non-anchoring sensitized semiconductor and the conventional sensitized semiconductor, Choi et al. sensitized TiO2 using an Sn porphyrin with pyridinyl and compared it with a ruthenium complex-sensitized TiO2 system. The SnP− species was detected by transient absorption spectroscopy and its lifetime was long enough to survive slow diffusion from the solution bulk to the surface of TiO2 (Fig. 12c), suggesting that SnTPyP could still sensitize TiO2/Pt to enhance photocatalytic activity in the pH 3–11 [169].
In conclusion, the strategy of non-contact sensitization can not only improve the operability of photocatalysts under different pH conditions but also apply more homogeneous porphyrin photocatalysts with a long lifetime of excited state to heterogeneous photocatalysis system. However, in this case, the carrier transport between the porphyrin derivative and the support depends on the diffusion of the porphyrin derivative in the solution and is limited by the diffusion coefficient. In addition, if porphyrin derivative is used as the active center of the target catalytic reaction, it will force us to consider the design of the selectivity and stability of photocatalytic reaction similar to the homogeneous catalytic system.
4. Structural design of porphyrin-organic frameworksBecause of the limited specific surface area of traditional inorganic photocatalysts, the mass loading of porphyrins is limited. In addition, the band structures of inorganic photocatalysts are difficult to be precisely regulated, which undoubtedly increases the difficulty in the design of heterogeneous junction between porphyrins and inorganic semiconductors.
Metal organic frameworks and covalent organic frameworks have a large specific surface area, precise structure, and easily adjustable energy band structure [170-175]. In addition, they can also be designed from bottom to top with the help of computational materials science (Fig. 13). Therefore, using the spatial confinement effect of the pores of the framework materials to adjust the electronic characteristics of the catalyst often produces unexpected effects.

|
Download:
|
| Fig. 13. Schematic diagram of the porphyrin-organic frameworks. | |
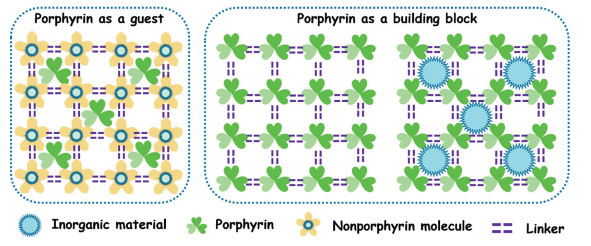
As we all know, the catalytic performance of frame materials mainly comes from the linkers, nodes, and the guests in the pores. It is easier to encapsulate the guests in the existing frame materials with rich and regular pores than to develop a new type of framework materials. At the same time, the properties of the guests and the frameworks can be well preserved. Since the porphyrin molecule has excellent light responsiveness and flexible active centers, encapsulating the porphyrin into the pores of the framework material can further increase the spectral response range and the number of active sites. Moreover, encapsulating the metal or carbon material into the frame material can increase the conductivity of the material [176-178].
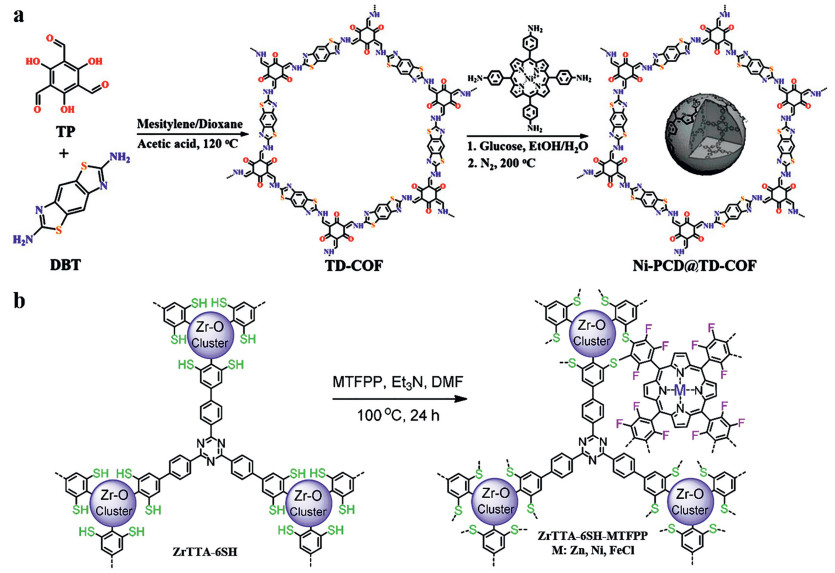
Wang et al. performed an ultrasound treatment of TD-COF with 5, 10, 15, 20-tetrakis(p-tetraphenylamino)porphyrin (Ni-TPAP) and glucose in solution, followed by selective pyrolysis process under N2 at 200 ℃ generated Ni-PCD@TD-COF (Fig. 14a), which contains metalloporphyrin-based carbon dots (M-PCD@TD-COF, M = Ni, Co, and Fe). The results showed a CO generated rate of 956 µmol/g in 2 h and the selectivity was as high as 98%. Although thermogravimetric analysis (TGA) demonstrated that the porphyrin structure was well preserved after the carbonization, almost no CO was produced without adding photosensitizer ([Ru(bpy)3]Cl2) [35].

|
Download:
|
| Fig. 14. (a) Schematic illustration for the synthesis of TD-COF and Ni-PCD@TD-COF. Reproduced with permission [35]. Copyright 2020, Wiley-VCH. (b) Schematic diagram of the covalent grafting process of porphyrin. Reproduced with permission [179]. Copyright 2020, American Chemical Society. | |
In order to use the photo-responsiveness of porphyrins and increase the metal content of organic framework materials, it is an ideal way to introduce porphyrins into frameworks in the form of molecules. However, these encapsulated small molecules and the framework material are often connected by non-covalent interactions [77-81], which limits the transfer of carriers.
To solve this problem, Xu et al. used the aromatic nucleophilic substitution between mercapto (-SH) and arylfluoro (Ar-F) groups to achieve the covalent graft of three different metalloporphyrins to metal-organic frameworks (Fig. 14b). The results showed that ZrTTA-6SH-ZnTFPP, ZrTTA-6SH-NiTFPP and ZrTTA-6SH-FeTFPP showed higher catalytic activity (110, 71 and 17 µmol g−1 h−1, respectively) than pristine ZrTTA-6SH. In this case, porphyrin photoexcited electrons can be transferred to Pt particles for hydrogen evolution reaction [179].
In addition, porphyrins can be used as mixed ligands to modify the metal-organic framework, which can also inhibit the aggregation of porphyrins and form robust connections. Wang et al. reported a one-pot synthetic strategy to chemically anchor Cu(Ⅱ) tetra(4-carboxylphenyl)porphyrin (CuTCPP) into UIO-66 MOF via a coordination method. XRD results showed that the crystal structure of UIO-66 was not damaged after the introduction of CuTCPP. Meanwhile, in-situ growth of TiO2 nanoparticles into the MOF was realized, which exhibited an optimal catalytic value of 31.32 µmol g−1 h−1 CO evolution rate (~7 times higher than that of pure TiO2) [180].
In a word, first of all, metalloporphyrins or metalloporphyrin composites are encapsulated in existing organic frameworks as guest materials, which can simply and effectively increase the active sites of hybrid photocatalysts. Secondly, the increased active sites can be combined with the excellent porous structure of organic frameworks to enhance the affinity of organic frameworks to substrates. Finally, in order to enhance the interaction between porphyrin guest and framework materials, and improve the activity and stability of photocatalyst, covalent grafting of molecules or using porphyrin as mixed ligand provides a new idea for the design of porphyrin-based organic frameworks.
4.2. Porphyrin as the building blocks in organic frameworksThe porphyrin guest occupying the pores of the framework material will reduce the specific surface area of the framework material, which is not conducive to the transportation of reactants and products. In order to solve this problem, researchers used porphyrin as a linking agent for organic framework blocks to maximize the excellent physical and chemical properties of porphyrin derivatives. In other words, by selecting appropriate substituents, the porphyrin is used as a building block to combine with other organic molecules or metal ions (or metal compounds) by covalent or coordination to form a covalent organic framework or metal organic framework (Table 2).
|
|
Table 2 Summary of catalytic properties of porphyrin-based organic photocatalysts reported in recent years. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jiang et al. reported a porphyrin-based metal organic framework (PCN-222, Fig. 15a), the results indicated that the formate was increasingly generated (30 µmol in 10 h), much higher than the values obtained over UiO-66-NH2, MIL-125-NH2 and the corresponding porphyrin ligand itself under similar conditions. The mechanism information was obtained from ultrafast transient absorption spectroscopy and time-resolved photoluminescence spectroscopy, elucidating that the detrimental electron-hole recombination was effectively inhibited in the presence of a deep electron trap state in PCN-222 [181].

|
Download:
|
| Fig. 15. (a) Schematic diagram of the photocatalytic process of PCN-222. Reproduced with permission [181]. Copyright 2015, American Chemical Society. (b) Proposed mechanism for the overall CO2 photoreduction of PCN-601. Reproduced with permission [39]. Copyright 2020, American Chemical Society. (c) Schematic of the mechanism of TTCOF-M CO2RR with H2O oxidation, theoretical simulation UV/vis DRS of TTCOF-Zn, and scheme of PET route under light excitation (inset). Reproduced with permission [183]. Copyright 2019, Wiley-VCH. | |
{kind=link}
For the metal-organic framework with carboxyl ligands as building blocks, there is a contradiction between activity and stability. In order to solve this problem, further reducing the harmful recombination of photogenerated carriers, improving the ligand-node charge transfer dynamics, shortening the ligand-node distance can improve the driving force of photogenerated carriers. Lin et al. synthesized two new metal-organic frameworks (Ru-TBP and Ru-TBP-Zn), through tetracarboxylate ligands of porphyrins and Ru2 secondary building units (SBUs). The distance from porphyrin ligands to Ru2 SBUs (~1.1 nm) shortens the transmission distance of optical carriers from porphyrins to Ru2 SBUs. Photocatalytic tests show that the H2 production rate increased linearly with time, and the rate of Ru-TBP-Zn was almost 2 times of that for Ru-TBP. Obviously, this did not fundamentally solve the contradiction between activity and selectivity, so it may be a wise choice to choose porphyrins containing other end-group groups as building blocks [182].
Zhou et al. synthesized a pyrazolyl porphyrinic Ni-MOF (PCN-601), which was different from MOF chelated by carboxylic acid. The pyrazolyl groups possess a larger π-conjugation system and cause higher π-d orbital overlaps with Ni-oxo nodes, which alleviates the tension between stability and activity. In addition, DFT calculations showed that photogenic electrons could be transferred from porphyrins to Ni oxo clusters, but not photogenic holes (Fig. 15b). This special band structure facilitated the carrier separation within the catalyst [39].
As for covalent organic frameworks using porphyrin derivatives as the linkers, how to improve the separation and transport of the carriers is also the key to enhance their photocatalytic performance. Lan et al. synthesized a series of Schiff base covalent organic framework (TTCOF-M, M = 2H, Zn, Ni, Cu) based on electron-deficient metalloporphyrin (TAPP) and electron-rich tetrathiafulvalene (TTF), the photocatalytic test showed that TTCOF-Zn achieved the best photocatalytic CO generation of 12.33 mmol with amazing selectivity (100%), along with generation O2. The interaction between electron donor and electron acceptor formed by TTF and TAPP provides a prerequisite for the regulation of metal centers in porphyrin rings. DFT calculations showed that HOMO and LUMO energy levels were distributed on TTF and TAPP respectively (Fig. 15c), which made the photogenic carriers with higher separation efficiency [183].
In general, compared with using porphyrin monomer as the photocatalyst, using porphyrin as a building block to construct an organic framework as photocatalyst can obtain more efficient photocatalytic efficiency. In addition, the band structure of organic frameworks can also be regulated by the end groups or metal centers of porphyrin derivatives. The easily constructed electron Donor-Acceptor junction, flexible active centers, and regular channels make porphyrin-based organic frameworks one of the favorites in the field of photocatalysis.
4.3. Application of spatial confinement effect in frame materialsIt is well known that the combination of inorganic semiconductors and porphyrin-based organic frameworks will be advantageous for further separation of the photocarriers [58-71, 184].
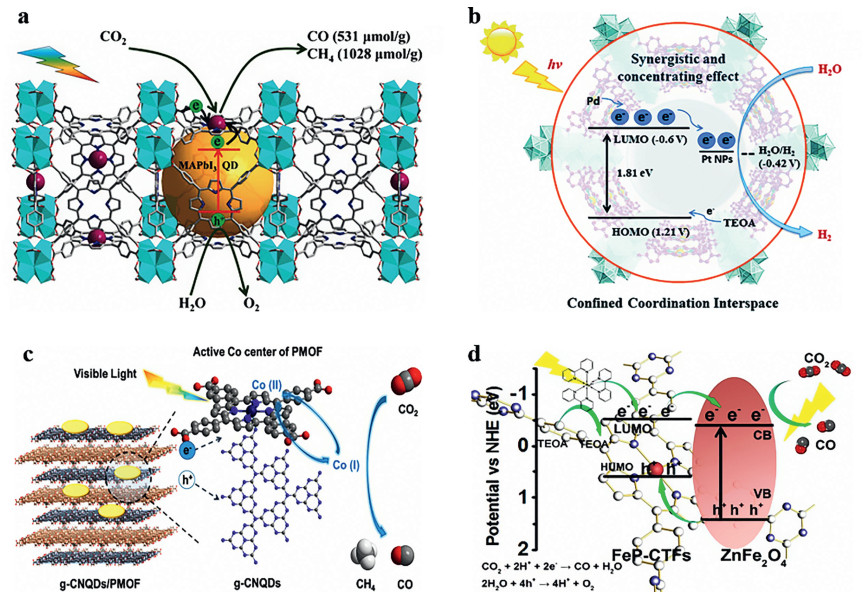
Lu et al. in-situ synthesized perovskite QDs in the orifice of PCN-221 (Fex, X = 0.2, 0.4, 0.6, 0.8, 1) through a continuous deposition route (Fig. 16a). The average size of quantum dots was about 1.8 nm and the aperture of PCN-221 was about 2 nm, so perovskite quantum dots can uniformly be confined into the PCN-221. The photocatalytic activity of the hybrid photocatalyst with added perovskite was significantly higher than that without adding perovskite. The subsequent TRPL test showed that perovskite QDs closely contact with Fe atoms in the PCN-221, leading to rapid charge transfer and obviously enhanced photocatalytic performances [185].

|
Download:
|
| Fig. 16. (a) Schematic diagram of the photocatalytic process of PCN-221(Fex)@MAPbI3 QDs. Reproduced with permission [185]. Copyright 2019 Wiley-VCH. (b) Schematic illustration of the structure and photocatalytic behavior of the MOF composite Pt@Pd-PCN-222(Hf). Reproduced with permission [186]. Copyright 2019 Royal Society of Chemistry. (c) The photocatalytic process of g-CNQDs/PMOF. Reproduced with permission [187]. Copyright 2019 American Chemical Society. (d) The proposed mechanism of the reduction of CO2 into CO photocatalyzed by ZnFe2O4/FeP-CTFs. Reproduced with permission [34]. Copyright 2020 Elsevier B.V. | |
{kind=link}
In addition, Su et al. used the in-situ reduction method to immobilize the platinum nanoparticles (Pt-NPs) with a width of about 3 nm into the confined coordination interspaces of 3.7 nm diameter, which are facilitated by early transition metal Hf(Ⅳ)-based clusters of a self-sensitized palladium porphyrin metal organic framework (Fig. 16b). The steady-state photoluminescence (PL) spectroscopy showed that Pt@Pd-PCN-222(Hf) had a lower photo-generated electron-hole recombination rate than Pd-PCN-222(Hf). Pt@Pd-PCN-222(Hf) had higher photocatalytic performance than that of unloaded Pt, and the performance did not decrease significantly in the three cycles, which further indicated that the Pt particles in the spatial limited domain had a better ability to prevent agglomeration [186].
Although the spatial confinement can effectively improve photocatalytic stability, the specific surface area of the hybrid material will be reduced. Therefore, it is another feasible method to encapsulate the host in-between the interlayers of the frame material. Recently, Liu et al. combined carbon nitride quantum dots with a two-dimensional porphyrin-based organic framework (Fig. 16c). Compared with previously reported hybrid catalysts via electrostatic or physical interactions, the g-CNQDs were coordinated with Co active sites in the PMOF, resulting in efficient separation of photogenerated charge carriers and effective injection of exciting electrons into Co centers for CO2 reduction. Notably, the prepared hybrid catalyst exhibited a 2.34-fold enhancement in CO generation rate (16.10 µmol g−1 h−1) and a 6.02-fold enhancement in the CH4 evolution rate (6.86 µmol g−1 h−1) compared to the bare PMOF [187].
In addition, the in-situ chemical deposition using the metal center of metallic porphyrin can further enhance the dispersivity and stability of the guest. Wang et al. reported a new and simple strategy for synthesizing composite ZnFe2O4/FeP-CTFs (Fig. 16d), where the Fe atoms coordinating to porphyrin rings of CTFs axially coordinated with ZnCl2 to form ZnFe2O4 and meanwhile loaded on the FeP-CTFs to form a heterojunction structure. Notably, a series of experiments elucidated that the anchoring sites of FeP-CTFs could effectively promote the crystal-plane ratio of high catalytic performance. Experimental measurements together with density functional theory calculations reveal that the strong interactions between ZnFe2O4 NPs and FeP-CTFs support can promote the photogenerated charge separation, thus facilitating photocatalytic CO2 reduction [34].
In conclusion, when the organic framework with porphyrin as a building block is combined with other materials, the encapsulated object will obtain better molecular dispersion and stability than macro-materials. In addition, for the guest with a certain crystal structure, it will show the preferred orientation of the crystal plane, which is very important to improve the activity and selectivity of the catalyst. Finally, the encapsulated guest often interacts with the active center of porphyrin, which provides a new way to adjust the performance of the photocatalyst.
5. Conclusions and perspectiveIn summary, porphyrin-based homogeneous photocatalysis has received extensive attention due to their higher activities and selectivities, but there are still some challenges that need to be solved such as recovery and poor stability. This review highlighted the recent progress of porphyrin-based heterogeneous photocatalysts. It is very important for molecular design by the modification of the end groups of the porphyrin molecule and the regulation of metal atoms in metalloporphyrins, thus improving the photo-responsiveness, selectivity, and stability of the photocatalyst. In order to further improve light absorption capacity and the separation efficiency of photogenerated carriers, as well as the recovery and stability of the porphyrin-based photocatalysts, strong interfacial covalent/noncovalent between porphyrins and inorganic or organic framework materials are mainly discussed.
Currently, although porphyrin-based heterogeneous photocatalysts have been used in many fields such as biomedicine, environmental governance, and energy conversion, they still face many challenges. First of all, in terms of the advantages and disadvantages of the porphyrin-based molecular catalysts, inorganic catalysts, and framework materials, a reasonable combination of two or more of these catalysts is still a challenge. Therefore, the consideration of the overall design of the photocatalytic system in the molecular and even electronic levels is needed such as the qualitative and quantitative rational configuration of light trapping components, carrier transport channels, and active centers. In addition, the photo-generated electrons and holes are produced under two half-reactions in the photocatalytic process, we should consider not only the recombination of photogenerated carriers based on different transfer mechanisms of electrons and holes but also the combination of the thermodynamic and kinetic processes of two different half-reactions for increasing the surface utilization of photogenerated carriers. In most porphyrin-based heterogeneous photocatalytic systems, the desired product in a half-reaction was mainly focused on, which not only increased the economic cost (sacrificial agent) but also caused a waste of energy. Therefore, effective utilization of the photogenerated electrons and holes at the same time remains a challenge. Finally, surface characteristics (such as hydrophilicity and hydrophobicity) of the photocatalysts should be considered in the different photocatalytic reactions (such as photocatalytic water splitting, photocatalytic reduction of carbon dioxide). Furthermore, a deep understanding of thermodynamics and kinetics of interfacial electron and hole transfer based on computational studies is beneficial for the design of an efficient catalytic system. We hope that this review will be helpful for the bottom-up design of porphyrin-based heterogeneous catalysts at the molecular and atomic levels, providing the photocatalytic technology to exploit solar energy for sustainable energy resources.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 51972345, 51972342), Taishan Scholar Project of Shandong Province (No. ts20190922), Key Basic Research Project of Natural Science Foundation of Shandong Province (No. ZR2019ZD51), Fundamental Research Funds for the Central Universities (No. 19CX05001A).
| [1] |
C.B. Kc, F. D'Souza, Coordin. Chem. Rev. 322 (2016) 104-141. DOI:10.1016/j.ccr.2016.05.012 |
| [2] |
X. Ji, J. Wang, L. Mei, et al., Adv. Funct. Mater. 28 (2018) 1705083. DOI:10.1002/adfm.201705083 |
| [3] |
X. Ji, J. Wang, Y. Kang, et al., ACS Catal. 8 (2018) 10732-10745. DOI:10.1021/acscatal.8b03105 |
| [4] |
G. Mukherjee, J. Thote, H.B. Aiyappa, et al., Chem. Commun. 53 (2017) 4461-4464. DOI:10.1039/C7CC00879A |
| [5] |
A.M. Huerta-Flores, G. Bengasi, K. Baba, N.D. Boscher, ACS Appl. Energy Mater. 3 (2020) 9848-9855. DOI:10.1021/acsaem.0c01545 |
| [6] |
K. Liu, C. Yuan, Q. Zou, Z. Xie, X. Yan, Angew. Chem. Int. Ed. 56 (2017) 7876-7880. DOI:10.1002/anie.201704678 |
| [7] |
R. Wang, C. He, W. Chen, C. Zhao, J. Huo, Chin. Chem. Lett. (2021). DOI:10.1016/j.cclet.2021.05.024 |
| [8] |
Y. Chen, S. Ji, W. Sun, et al., Angew. Chem. Int. Ed. 59 (2020) 1295-1301. DOI:10.1002/anie.201912439 |
| [9] |
P. Fu, S. Hu, J. Tang, Z. Xiao, Front. Optoelectronics (2021). DOI:10.1007/s12200-021-1227-z |
| [10] |
L. Chen, Y. Song, Y. Liu, et al., J. Energ. Chem. 50 (2020) 395-401. DOI:10.1016/j.jechem.2020.03.046 |
| [11] |
Y. Lei, Y. Wang, Y. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 20794-20812. DOI:10.1002/anie.201914647 |
| [12] |
Y. Liu, Q. Feng, W. Liu, et al., Nano Energy 81 (2021) 105641. DOI:10.1016/j.nanoen.2020.105641 |
| [13] |
J. Wang, C. He, J. Huo, L. Fu, C. Zhao, Adv. Theory Simul. 4 (2021) 2100003. DOI:10.1002/adts.202100003 |
| [14] |
J. Yu, C. He, C. Pu, et al., Chin. Chem. Lett. 32 (2021) 3149-3154. DOI:10.1016/j.cclet.2021.02.046 |
| [15] |
H. Yang, C. He, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3202-3206. DOI:10.1016/j.cclet.2021.03.038 |
| [16] |
W. Song, J. Wang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3137-3142. DOI:10.1016/j.cclet.2021.02.043 |
| [17] |
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J. 414 (2021) 128857. DOI:10.1016/j.cej.2021.128857 |
| [18] |
X. Chang, T. Wang, J. Gong, Energy Environ. Sci. 9 (2016) 2177-2196. DOI:10.1039/C6EE00383D |
| [19] |
W. Tu, Y. Zhou, Z. Zou, Adv. Mater. 26 (2014) 4607-4626. DOI:10.1002/adma.201400087 |
| [20] |
H. Kotani, T. Miyazaki, E. Aoki, et al., ACS Appl. Energy Mater. 3 (2020) 3193-3197. DOI:10.1021/acsaem.0c00206 |
| [21] |
M. Watanabe, S. Sun, T. Ishihara, et al., ACS Appl. Energy Mater. 1 (2018) 6072-6081. DOI:10.1021/acsaem.8b01113 |
| [22] |
S. Gonuguntla, A. Tiwari, S. Madanaboina, G. Lingamallu, U. Pal, Int. J. Hydrogen. Energ 45 (2020) 7508-7516. DOI:10.1016/j.ijhydene.2019.04.268 |
| [23] |
E. Koposova, X. Liu, A. Pendin, et al., J. Phys. Chem. C. 120 (2016) 13873-13890. DOI:10.1021/acs.jpcc.6b01467 |
| [24] |
R. Ge, X. Li, B. Zhuang, et al., Appl. Catal., B 211 (2017) 296-304. DOI:10.1016/j.apcatb.2017.04.056 |
| [25] |
K. Li, L. Lin, T. Peng, et al., Chem. Commun. 51 (2015) 12443-12446. DOI:10.1039/C5CC03812J |
| [26] |
H. Gao, J. Wang, M. Jia, et al., Chem. Eng. J. 374 (2019) 684-693. DOI:10.1016/j.cej.2019.06.002 |
| [27] |
X. Ning, Y. Wu, X. Ma, et al., Adv. Funct. Mater. 29 (2019) 1902992. DOI:10.1002/adfm.201902992 |
| [28] |
P. Li, X. Zhang, C. Hou, Y. Chen, T. He, Appl. Catal. B 238 (2018) 656-663. DOI:10.1016/j.apcatb.2018.07.066 |
| [29] |
S. Mei, J. Gao, Y. Zhang, et al., J. Colloid Interface Sci. 506 (2017) 58-65. DOI:10.1016/j.jcis.2017.07.030 |
| [30] |
G. Zhao, H. Pang, G. Liu, et al., Appl. Catal. B 200 (2017) 141-149. DOI:10.1016/j.apcatb.2016.06.074 |
| [31] |
L. Lin, C. Hou, X. Zhang, et al., Appl. Catal. B 221 (2018) 312-319. DOI:10.1016/j.apcatb.2017.09.033 |
| [32] |
S. Tian, S. Chen, X. Ren, et al., Nano Res. 13 (2020) 2665-2672. DOI:10.1007/s12274-020-2908-4 |
| [33] |
N. Xu, Y. Diao, X. Qin, et al., Dalton. Trans. 49 (2020) 15587-15591. DOI:10.1039/D0DT03205K |
| [34] |
Yan Y.l., Q.J. Fang, J.K. Pan, et al., Chem. Eng. J. 408 (2021) 127358. DOI:10.1016/j.cej.2020.127358 |
| [35] |
H. Zhong, R. Sa, H. Lv, et al., Adv. Funct. Mater. 30 (2020) 2002654. DOI:10.1002/adfm.202002654 |
| [36] |
F. Leng, H. Liu, M. Ding, Q.P. Lin, H.L. Jiang, ACS Catal. 8 (2018) 4583-4590. DOI:10.1021/acscatal.8b00764 |
| [37] |
J. Liu, Y.Z. Fan, X. Li, et al., Appl. Catal. B 231 (2018) 173-181. DOI:10.1016/j.apcatb.2018.02.055 |
| [38] |
N. Sadeghi, S. Sharifnia, T.Q. Do, J. Mater. Chem. A 6 (2018) 18031-18035. DOI:10.1039/C8TA07158F |
| [39] |
Z.B. Fang, T.T. Liu, J. Liu, et al., J. Am. Chem. Soc. 142 (2020) 12515-12523. DOI:10.1021/jacs.0c05530 |
| [40] |
B. Mondal, A. Rana, P. Sen, A. Dey, J. Am. Chem. Soc. 137 (2015) 11214-11217. DOI:10.1021/jacs.5b05992 |
| [41] |
X. Zhang, M. Cibian, A. Call, K. Yamauchi, K. Sakai, ACS Catal. 9 (2019) 11263-11273. DOI:10.1021/acscatal.9b04023 |
| [42] |
Y. Kuramochi, Y. Fujisawa, A. Satake, J. Am. Chem. Soc. 142 (2020) 705-709. DOI:10.1021/jacs.9b12712 |
| [43] |
Z. Fan, K. Nomura, M. Zhu, et al., Commun. Chem. 2 (2019) 55. DOI:10.1038/s42004-019-0158-8 |
| [44] |
F. Kuttassery, S. Sagawa, S. Mathew, et al., ACS Appl. Energy Mater. 2 (2019) 8045-8051. DOI:10.1021/acsaem.9b01552 |
| [45] |
X.F. Liu, R.X. Li, X.T. Ren, et al., J. Catal. 348 (2017) 314-320. DOI:10.1016/j.jcat.2016.12.014 |
| [46] |
Z. Ma, Q. Zhang, S. Panda, et al., Sustain. Energy Fuels 4 (2020) 4694-4703. DOI:10.1039/D0SE00818D |
| [47] |
M. Zhu, Y. Du, P. Yang, X. Wang, Catal. Sci. Technol. 3 (2013) 2295-2302. DOI:10.1039/c3cy00236e |
| [48] |
H. Imahori, T. Umeyama, S. Ito, Accounts. Chem. Res. 42 (2009) 1809-1818. DOI:10.1021/ar900034t |
| [49] |
L.J. Wang, R.L. Wang, X. Zhang, et al., ChemSusChem 13 (2020) 2973-2980. DOI:10.1002/cssc.202000103 |
| [50] |
L. Jin, S. Lv, Y. Miao, D. Liu, F. Song, ChemCatChem 13 (2021) 140-152. DOI:10.1002/cctc.202001179 |
| [51] |
R.K. Yadav, J.-O. Baeg, G.H. Oh, et al., J. Am. Chem. Soc. 134 (2012) 11455-11461. DOI:10.1021/ja3009902 |
| [52] |
A. Call, M. Cibian, K. Yamamoto, et al., ACS Catal. 9 (2019) 4867-4874. DOI:10.1021/acscatal.8b04975 |
| [53] |
P. Lang, M. Pfrunder, G. Quach, et al., Chem. Eur. J. 25 (2019) 4509-4519. DOI:10.1002/chem.201806347 |
| [54] |
Y.Q. Zhang, J.Y. Chen, P.E.M. Siegbahn, R.Z. Liao, ACS Catal. 10 (2020) 6332-6345. DOI:10.1021/acscatal.0c00559 |
| [55] |
H. Rao, J. Bonin, M. Robert, Chem. Commun. 53 (2017) 2830-2833. DOI:10.1039/C6CC09967J |
| [56] |
M. Natali, R. Argazzi, C. Chiorboli, E. Iengo, F. Scandola, Chem. Eur. J. 19 (2013) 9261-9271. DOI:10.1002/chem.201300133 |
| [57] |
A.M. Kluwer, R. Kapre, F. Hartl, et al., Proc. Natl. Acad. Sci. 106 (2009) 10460-10466. DOI:10.1073/pnas.0809666106 |
| [58] |
K. Matsuyama, M. Motomura, T. Kato, T. Okuyama, H. Muto, Micropor. Mesopor. Mater. 225 (2016) 26-32. DOI:10.1016/j.micromeso.2015.12.005 |
| [59] |
Z. Zheng, H. Xu, Z. Xu, J. Ge, Small 14 (2018) 1702812. DOI:10.1002/smll.201702812 |
| [60] |
Y. Huang, Y. Zhang, X. Chen, et al., Chem. Commun. 50 (2014) 10115-10117. DOI:10.1039/C4CC04479G |
| [61] |
Z. Ding, K. Wang, Z. Mai, et al., Int. J. Hydrogen. Energ 44 (2019) 24680-24689. DOI:10.1016/j.ijhydene.2019.07.244 |
| [62] |
Y.-H. Zhou, X. Cao, J. Ning, et al., Int. J. Hydrogen. Energ 45 (2020) 31440-31451. DOI:10.1016/j.ijhydene.2020.08.141 |
| [63] |
L. Chang, Y. Li, Mol. Catal. 433 (2017) 77-83. DOI:10.1016/j.mcat.2017.01.009 |
| [64] |
X. Yang, Q. Xu, Trends Chem. 2 (2020) 214-226. DOI:10.1016/j.trechm.2019.12.001 |
| [65] |
N. Zhang, Q. Shao, P. Wang, X. Zhu, X. Huang, Small 14 (2018) 1704318. DOI:10.1002/smll.201704318 |
| [66] |
R.J. Young, M.T. Huxley, E. Pardo, et al., Chem. Sci. 11 (2020) 4031-4050. DOI:10.1039/D0SC00485E |
| [67] |
W. Cheng, Y. Peng, Y. Wang, et al., Int. J. Hydrogen. Energ 46 (2021) 2204-2212. DOI:10.1016/j.ijhydene.2020.10.140 |
| [68] |
C. Xu, J. Lin, D. Yan, et al., ACS Appl. Nano Mater. 3 (2020) 6416-6422. DOI:10.1021/acsanm.0c00884 |
| [69] |
L. Chen, B. Huang, X. Qiu, et al., Chem. Sci. 7 (2016) 228-233. DOI:10.1039/C5SC02925B |
| [70] |
J. Cure, E. Mattson, K. Cocq, et al., J. Mater. Chem. A 7 (2019) 17536-17546. DOI:10.1039/C8TA12334A |
| [71] |
N. Cao, S. Tan, W. Luo, K. Hu, G. Cheng, Catal. Lett. 146 (2016) 518-524. DOI:10.1007/s10562-015-1671-8 |
| [72] |
L. Li, H. Zhao, J. Wang, R. Wang, ACS Nano 8 (2014) 5352-5364. DOI:10.1021/nn501853g |
| [73] |
Z. Li, H. Zhang, L. Kong, et al., J. Environ. Chem. Eng. 8 (2020) 104363. DOI:10.1016/j.jece.2020.104363 |
| [74] |
B. An, J. Zhang, K. Cheng, et al., J. Am. Chem. Soc. 139 (2017) 3834-3840. DOI:10.1021/jacs.7b00058 |
| [75] |
J. Wei, Y. Chen, H. Zhang, Z. Zhuang, Y. Yu, Chin. J. Catal. 42 (2021) 78-86. DOI:10.1016/S1872-2067(20)63661-0 |
| [76] |
S. Laha, D. Rambabu, S. Bhattacharyya, T.K. Maji, Chem. Eur. J. 26 (2020) 14671-14678. DOI:10.1002/chem.202002439 |
| [77] |
J. Yang, H. Guo, S. Chen, et al., J. Mater. Chem. A 6 (2018) 13859-13866. DOI:10.1039/C8TA03249A |
| [78] |
L. Tang, J. Shi, H. Wu, et al., Nanotechnology 28 (2017) 365604. DOI:10.1088/1361-6528/aa79e1 |
| [79] |
X. Zhao, H. Xu, X. Wang, et al., ACS Appl. Mater. Interfaces 10 (2018) 15096-15103. DOI:10.1021/acsami.8b03561 |
| [80] |
Y. Yue, A.J. Binder, R. Song, et al., Dalton. Trans. 43 (2014) 17893-17898. DOI:10.1039/C4DT02516D |
| [81] |
L. Ma, W. Hu, B. Mei, et al., ACS Catal. 10 (2020) 4534-4542. DOI:10.1021/acscatal.0c00243 |
| [82] |
T. Zhao, I. Boldog, V. Spasojevic, et al., J. Mater. Chem. C 4 (2016) 6588-6601. DOI:10.1039/C6TC01297C |
| [83] |
R. Ren, H. Zhao, X. Sui, et al., Catalysts 9 (2019) 89. DOI:10.3390/catal9010089 |
| [84] |
D.I. Won, J.S. Lee, Q. Ba, et al., ACS Catal. 8 (2018) 1018-1030. DOI:10.1021/acscatal.7b02961 |
| [85] |
S. Lian, M.S. Kodaimati, E.A. Weiss, ACS Nano 12 (2018) 568-575. DOI:10.1021/acsnano.7b07377 |
| [86] |
Z. Zhang, Y. Zhu, X. Chen, H. Zhang, J. Wang, Adv. Mater. 31 (2019) 1806626. DOI:10.1002/adma.201806626 |
| [87] |
G.B. Bodedla, J. Huang, W.-Y. Wong, X. Zhu, ACS Appl. Nano Mater. 3 (2020) 7040-7046. |
| [88] |
N. Zhang, L. Wang, H. Wang, et al., Nano Lett. 18 (2018) 560-566. DOI:10.1021/acs.nanolett.7b04701 |
| [89] |
G. Yang, C. Lin, X. Feng, T. Wang, J. Jiang, Chem. Commun. 56 (2020) 527-530. DOI:10.1039/C9CC08060K |
| [90] |
J. Wang, Y. Zhong, L. Wang, et al., Nano Lett. 16 (2016) 6523-6528. DOI:10.1021/acs.nanolett.6b03135 |
| [91] |
H.H. Tsai, M.C. Simpson, Chem. Phys. Lett. 353 (2002) 111-118. DOI:10.1016/S0009-2614(01)01457-9 |
| [92] |
L.L. Li, E.W.G. Diau, Chem. Soc. Rev. 42 (2013) 291-304. DOI:10.1039/C2CS35257E |
| [93] |
R. Frisenda, A.J. Molina-Mendoza, T. Mueller, A. Castellanos-Gomez, H.S.J. van der Zant, Chem. Soc. Rev. 47 (2018) 3339-3358. DOI:10.1039/C7CS00880E |
| [94] |
X. Zou, L. Ji, X. Yang, et al., J. Am. Chem. Soc. 139 (2017) 16060-16063. DOI:10.1021/jacs.7b09090 |
| [95] |
Y. Zhang, R. Suzuki, Y. Iwasa, ACS Nano 11 (2017) 12583-12590. DOI:10.1021/acsnano.7b06752 |
| [96] |
G. Pu, Z. Yang, Y. Wu, et al., Anal. Chem. 91 (2019) 2319-2328. DOI:10.1021/acs.analchem.8b05027 |
| [97] |
E.S. Da Silva, N.M.M. Moura, M.G.P.M.S. Neves, et al., Appl. Catal. 221 (2018) 56-69. DOI:10.1016/j.apcatb.2017.08.079 |
| [98] |
T. Higashino, Y. Kurumisawa, S. Nimura, H. Iiyama, H. Imahori, Eur. J. Org. Chem. 2018 (2018) 2537-2547. DOI:10.1002/ejoc.201701736 |
| [99] |
J. Nieto-Pescador, B. Abraham, J. Li, et al., J. Phys. Chem. C. 120 (2016) 48-55. DOI:10.1021/acs.jpcc.5b09463 |
| [100] |
K.J. Shah, T. Imae, J. Mater. Chem. A 5 (2017) 9691-9701. DOI:10.1039/C7TA01861D |
| [101] |
L. Xie, J. Tian, Y. Ouyang, et al., Angew. Chem. Int. Ed. 59 (2020) 15844-15848. DOI:10.1002/anie.202003836 |
| [102] |
Y. Zhang, Y. Wang, L.C. An, et al., Mater. Chem. Front. 4 (2020) 2754-2761. DOI:10.1039/D0QM00314J |
| [103] |
J. Wang, Y. Zheng, T. Peng, J. Zhang, R. Li, ACS Sustain. Chem. Eng. 5 (2017) 7549-7556. DOI:10.1021/acssuschemeng.7b00700 |
| [104] |
P. Zeng, Y. Zheng, S. Chen, et al., New J. Chem. 44 (2020) 11237-11247. DOI:10.1039/D0NJ02056G |
| [105] |
M.J. Latter, S.J. Langford, Int. J. Mol. Sci. 11 (2010) 1878-1887. DOI:10.3390/ijms11041878 |
| [106] |
E. Barton Cole, P.S. Lakkaraju, D.M. Rampulla, et al., J. Am. Chem. Soc. 132 (2010) 11539-11551. DOI:10.1021/ja1023496 |
| [107] |
M. Imran, M. Ramzan, A.K. Qureshi, M.A. Khan, M. Tariq, Biosensors 8 (2018) 95. DOI:10.3390/bios8040095 |
| [108] |
Y. Liu, L. Wang, H. Feng, et al., Nano Lett. 19 (2019) 2614-2619. DOI:10.1021/acs.nanolett.9b00423 |
| [109] |
S.S. Wang, H.H. Huang, M. Liu, et al., Inorg. Chem. 59 (2020) 6301-6307. DOI:10.1021/acs.inorgchem.0c00407 |
| [110] |
S. Fukuzumi, T. Kojima, J. Mater. Chem. 18 (2008) 1427-1439. DOI:10.1039/b717958h |
| [111] |
M.S. Choi, T. Yamazaki, I. Yamazaki, T. Aida, Angew. Chem. Int. Ed. 43 (2004) 150-158. DOI:10.1002/anie.200301665 |
| [112] |
M.N. Ha, G. Lu, Z. Liu, L. Wang, Z. Zhao, J. Mater. Chem. A 4 (2016) 13155-13165. DOI:10.1039/C6TA05402A |
| [113] |
A. Fateeva, P.A. Chater, C.P. Ireland, et al., Angew. Chem. Int. Ed. 51 (2012) 7440-7444. DOI:10.1002/anie.201202471 |
| [114] |
Y. Liu, Y. Yang, Q. Sun, et al., ACS Appl. Mater. Interfaces 5 (2013) 7654-7658. DOI:10.1021/am4019675 |
| [115] |
R.N. Sampaio, D.C. Grills, D.E. Polyansky, D.J. Szalda, E. Fujita, J. Am. Chem. Soc. 142 (2020) 2413-2428. DOI:10.1021/jacs.9b11897 |
| [116] |
X. Wang, Z. Chen, X. Zhao, et al., Angew. Chem. Int. Ed. 57 (2018) 1944-1948. DOI:10.1002/anie.201712451 |
| [117] |
Z. Chen, J. Wang, S. Zhang, et al., ACS Appl. Energy Mater. 2 (2019) 5665-5676. DOI:10.1021/acsaem.9b00811 |
| [118] |
Y.N. Gong, W. Zhong, Y. Li, et al., J. Am. Chem. Soc. 142 (2020) 16723-16731. DOI:10.1021/jacs.0c07206 |
| [119] |
A. Fujishima, K. Honda, Nature 238 (1972) 37-38. DOI:10.1038/238037a0 |
| [120] |
Y. Yuan, H. Lu, Z. Ji, et al., Chem. Eng. J. 275 (2015) 8-16. DOI:10.1016/j.cej.2015.04.015 |
| [121] |
Y.J. Yuan, J.R. Tu, Z.J. Ye, et al., Dyes Pigments 123 (2015) 285-292. DOI:10.1016/j.dyepig.2015.08.014 |
| [122] |
Y.J. Yuan, D. Chen, J. Zhong, et al., J. Phys. Chem. C 121 (2017) 24452-24462. DOI:10.1021/acs.jpcc.7b08290 |
| [123] |
P. Li, C. Hou, X. Zhang, Y. Chen, T. He, Appl. Surf. Sci. 459 (2018) 292-299. DOI:10.1016/j.apsusc.2018.08.002 |
| [124] |
K. Liu, R. Xing, Y. Li, et al., Angew. Chem. Int. Ed. 55 (2016) 12503-12507. DOI:10.1002/anie.201606795 |
| [125] |
F. Akbari Beni, A. Gholami, A. Ayati, M. Niknam Shahrak, M. Sillanpää, Micropor. Mesopor. Mater. 303 (2020) 110275. DOI:10.1016/j.micromeso.2020.110275 |
| [126] |
E.V. Ramos-Fernandez, C. Pieters, B. van der Linden, et al., J. Catal. 289 (2012) 42-52. DOI:10.1016/j.jcat.2012.01.013 |
| [127] |
B. Yan, K. Qian, Photochem. Photobiol. 85 (2009) 1278-1285. DOI:10.1111/j.1751-1097.2009.00596.x |
| [128] |
B. Yan, H.F. Lu, Inorg. Chem. 47 (2008) 5601-5611. DOI:10.1021/ic7021825 |
| [129] |
M. Guo, B. Yan, L. Guo, X.F. Qiao, Colloids Surf. A 380 (2011) 53-59. DOI:10.1016/j.colsurfa.2011.02.018 |
| [130] |
D.M. Fabian, A.M. Ganose, J.W. Ziller, et al., ACS Appl. Energy Mater. 2 (2019) 1579-1587. |
| [131] |
F.D. Lewis, J. Liu, W. Weigel, et al., Proc. Natl. Acad. Sci. 99 (2002) 12536-12541. DOI:10.1073/pnas.192432899 |
| [132] |
P. Urchaga, M. Weissmann, S. Baranton, T. Girardeau, C. Coutanceau, Langmuir 25 (2009) 6543-6550. DOI:10.1021/la9000973 |
| [133] |
C. Schubert, J.T. Margraf, T. Clark, D.M. Guldi, Chem. Soc. Rev. 44 (2015) 988-998. DOI:10.1039/C4CS00262H |
| [134] |
C. Benesch, M.F. Rode, M. Čízĕk, et al., J. Phys. Chem. C 113 (2009) 10315-10318. |
| [135] |
P. Wang, M. Xi, S.Z. Kang, et al., Int. J. Hydrogen. Energ 45 (2020) 6508-6518. DOI:10.1016/j.ijhydene.2019.12.179 |
| [136] |
X. Guo, X. Li, L. Qin, S.Z. Kang, G. Li, Appl. Catal. B 243 (2019) 1-9. DOI:10.1016/j.apcatb.2018.10.030 |
| [137] |
M. Xi, P. Wang, M. Zhang, et al., Appl. Surf. Sci. 529 (2020) 147200. DOI:10.1016/j.apsusc.2020.147200 |
| [138] |
L. Li, G.B. Bodedla, Z. Liu, X. Zhu, Appl. Surf. Sci. 499 (2020) 143755. DOI:10.1016/j.apsusc.2019.143755 |
| [139] |
S. Tian, S. Chen, X. Ren, et al., Nano Res. 12 (2019) 3109-3115. DOI:10.1007/s12274-019-2562-x |
| [140] |
Q. Luo, R. Ge, S.Z. Kang, et al., Appl. Surf. Sci. 427 (2018) 15-23. DOI:10.1016/j.apsusc.2017.08.152 |
| [141] |
M. Zhu, Z. Li, B. Xiao, et al., ACS Appl. Mater. Interfaces 5 (2013) 1732-1740. DOI:10.1021/am302912v |
| [142] |
X. Zhang, L. Lin, D. Qu, et al., Appl. Catal. B 265 (2020) 118595. DOI:10.1016/j.apcatb.2020.118595 |
| [143] |
L. Wang, S. Duan, P. Jin, et al., Appl. Catal. B 239 (2018) 599-608. DOI:10.1016/j.apcatb.2018.08.007 |
| [144] |
Q. Li, Q. Zeng, L. Gao, et al., J. Mater. Chem. A 5 (2017) 155-164. DOI:10.1039/C6TA07508H |
| [145] |
T. Hasobe, H. Sakai, K. Mase, K. Ohkubo, S. Fukuzumi, J. Phys. Chem. C 117 (2013) 4441-4449. DOI:10.1021/jp400381h |
| [146] |
R. Ge, X. Li, S.Z. Kang, L. Qin, G. Li, Appl. Catal. B 187 (2016) 67-74. DOI:10.1016/j.apcatb.2016.01.024 |
| [147] |
K. Zhu, Q. Luo, S.Z. Kang, et al., Nanoscale 10 (2018) 18635-18641. DOI:10.1039/C8NR06138F |
| [148] |
Z. Zou, J. Ye, H. Arakawa, J. Phys. Chem. B 106 (2002) 517-520. DOI:10.1021/jp012982f |
| [149] |
U. Alam, A. Khan, D. Ali, D. Bahnemann, M. Muneer, RSC Adv. 8 (2018) 17582-17594. DOI:10.1039/C8RA01638K |
| [150] |
N.U. Saqib, R. Adnan, I. Shah, Environ. Sci. Pollut. Res. 23 (2016) 15941-15951. DOI:10.1007/s11356-016-6984-7 |
| [151] |
L. Zhang, L. Qin, S.Z. Kang, G.D. Li, X. Li, ACS Sustain. Chem. Eng. 7 (2019) 8358-8366. DOI:10.1021/acssuschemeng.8b06867 |
| [152] |
K. Zhu, M. Zhang, X. Feng, et al., Appl. Catal. B 268 (2020) 118434. DOI:10.1016/j.apcatb.2019.118434 |
| [153] |
M. Zhang, K. Zhu, L. Qin, A.Z. Kang, X. Li, Catal. Sci. Technol. 10 (2020) 1640-1649. DOI:10.1039/C9CY02272D |
| [154] |
K. Ohkubo, P.J. Sintic, N.V. Tkachenko, et al., Chem. Phys. 326 (2006) 3-14. DOI:10.1016/j.chemphys.2006.01.034 |
| [155] |
Z. Xia, N. Xue, W. Shi, C. Lu, J. Phys. Chem. C 123 (2019) 30536-30544. DOI:10.1021/acs.jpcc.9b08488 |
| [156] |
S. Fukuzumi, K. Ohkubo, T. Suenobu, Accounts. Chem. Res. 47 (2014) 1455-1464. DOI:10.1021/ar400200u |
| [157] |
K. Ohkubo, S. Fukuzumi, B. Chem, Soc. Jpn. 82 (2009) 303-315. |
| [158] |
N.L. Bill, M. Ishida, Y. Kawashima, et al., Chem. Sci. 5 (2014) 3888-3896. DOI:10.1039/C4SC00803K |
| [159] |
J.B. Kelber, N.A. Panjwani, D. Wu, et al., Chem. Sci. 6 (2015) 6468-6481. DOI:10.1039/C5SC01830G |
| [160] |
K. Sakakibara, S. Nagino, H. Akanuma, et al., Dalton. Trans. 46 (2017) 12645-12654. DOI:10.1039/C7DT02674A |
| [161] |
M. Natali, A. Amati, N. Demitri, E. Iengo, Chem. Commun. 54 (2018) 6148-6152. DOI:10.1039/C8CC03441A |
| [162] |
K. Ohkubo, R. Garcia, P.J. Sintic, et al., Chem. Eur. J. 15 (2009) 10493-10503. DOI:10.1002/chem.200901105 |
| [163] |
P.K. Poddutoori, N. Zarrabi, A.G. Moiseev, et al., Chem. Eur. J. 19 (2013) 3148-3161. DOI:10.1002/chem.201202995 |
| [164] |
M.S. Asano, M. Shibuki, T. Otsuka, Chem. Lett. 45 (2016) 1114-1116. DOI:10.1246/cl.160442 |
| [165] |
B.W. Cohen, B.M. Lovaasen, C.K. Simpson, et al., Inorg. Chem. 49 (2010) 5777-5779. DOI:10.1021/ic100316s |
| [166] |
H. Imahori, D.M. Guldi, K. Tamaki, et al., J. Am. Chem. Soc. 123 (2001) 6617-6628. DOI:10.1021/ja004123v |
| [167] |
S. Fukuzumi, K. Ohkubo, et al., J. Am. Chem. Soc. 125 (2003) 14984-14985. DOI:10.1021/ja037214b |
| [168] |
J. Li, Y. Qiao, T. Pan, et al., Sensors 18 (2018) 3752. |
| [169] |
W. Kim, T. Tachikawa, T. Majima, et al., Energy Environ. Sci. 3 (2010) 1789-1795. DOI:10.1039/c0ee00205d |
| [170] |
Q. Zuo, T. Liu, C. Chen, et al., Angew. Chem. Int. Ed. 58 (2019) 10198-10203. DOI:10.1002/anie.201904058 |
| [171] |
E.X. Chen, M. Qiu, Y.F. Zhang, et al., Adv. Mater. 30 (2018) 1704388. DOI:10.1002/adma.201704388 |
| [172] |
Y. Hou, E. Zhang, J. Gao, et al., Dalton. Trans. 49 (2020) 7592-7597. DOI:10.1039/D0DT01436B |
| [173] |
L. Chen, Y. Wang, F. Yu, X. Shen, C. Duan, J. Mater. Chem. A 7 (2019) 11355-11361. DOI:10.1039/C9TA01840A |
| [174] |
N. Sadeghi, S. Sharifnia, M. Sheikh Arabi, J. CO2 Util. 16 (2016) 450-457. DOI:10.1016/j.jcou.2016.10.006 |
| [175] |
H. Zhang, J. Wei, J. Dong, et al., Angew. Chem. Int. Ed. 55 (2016) 14310-14314. DOI:10.1002/anie.201608597 |
| [176] |
H.G.R. Monestel, I.S. Amiinu, A.A. González, et al., Chin. J. Catal. 41 (2020) 839-846. DOI:10.1016/S1872-2067(19)63488-1 |
| [177] |
Y. Li, X. Cai, S. Chen, et al., ChemSusChem 11 (2018) 1040-1047. DOI:10.1002/cssc.201800016 |
| [178] |
Y.H. Xiao, W. Tian, S. Jin, Z.G. Gu, J. Zhang, Small 16 (2020) 2005111. DOI:10.1002/smll.202005111 |
| [179] |
Y. Diao, N. Xu, M.Q. Li, X. Zhu, Z. Xu, Inorg. Chem. 59 (2020) 12643-12649. DOI:10.1021/acs.inorgchem.0c01744 |
| [180] |
L. Wang, P. Jin, S. Duan, et al., Sci. Bull. 64 (2019) 926-933. DOI:10.1016/j.scib.2019.05.012 |
| [181] |
H.Q. Xu, J. Hu, D. Wang, et al., J. Am. Chem. Soc. 137 (2015) 13440-13443. DOI:10.1021/jacs.5b08773 |
| [182] |
G. Lan, Y.-Y. Zhu, S.S. Veroneau, et al., J. Am. Chem. Soc. 140 (2018) 5326-5329. DOI:10.1021/jacs.8b01601 |
| [183] |
M. Lu, J. Liu, Q. Li, et al., Angew. Chem. Int. Ed. 58 (2019) 12392-12397. DOI:10.1002/anie.201906890 |
| [184] |
X. Yang, J.K. Sun, M. Kitta, H. Pang, Q. Xu, Nat. Catal. 1 (2018) 214-220. DOI:10.1038/s41929-018-0030-8 |
| [185] |
L.Y. Wu, Y.F. Mu, X.X. Guo, et al., Angew. Chem. Int. Ed. 58 (2019) 9491-9495. DOI:10.1002/anie.201904537 |
| [186] |
S. Li, H.M. Mei, S.L. Yao, et al., Chem. Sci. 10 (2019) 10577-10585. DOI:10.1039/C9SC01866B |
| [187] |
C. Zheng, X. Qiu, J. Han, Y. Wu, S. Liu, ACS Appl. Mater. Interfaces 11 (2019) 42243-42249. DOI:10.1021/acsami.9b15306 |