 2022, Vol. 33
2022, Vol. 33 


b Textile Pollution Controlling Engineering Center of Ministry of Environmental Protection, College of Environmental Science and Engineering, Donghua University, Shanghai 201620, China
Water pollution is an environmental issue of global concern, usually resulting from the human activities. Consequently, the contamination of water bodies is expected to increase with increasing industrialization and population growth [1]. In particular, the increased incidence of emerging organic contaminants (EOCs; e.g., flame-retardants, life-style compounds, pharmaceuticals and personal care products, pesticides and their degradation products pharmaceuticals, surfactants) in groundwater from urban and agricultural sources [2, 3], previously undetectable, is of concern as specialized treatment technologies may be required for their removal [4]. Studies have shown that conventional treatment technologies, such as adsorption, flocculation, ultrafiltration, and biological oxidation, may be ineffective for the removal or degradation of these contaminants due to their slow reaction kinetics and poor selectivity [5].
Advanced oxidation processes (AOPs) are expedient treatment technology involving diverse reactive oxygen species (ROS) for the detoxification of contaminants in water systems [6-9]. They have the compelling advantage of completely mineralizing organic pollutants instead of simply isolating them from contaminated waters [10, 11]. Among these AOPs, persulfate-based treatments have received considerable attention in recent years due to their strong oxidative capabilities and wide pH tolerance [12, 13]. Compared to conventional AOPs in which hydroxyl radicals (•OH) serve as the main oxidant, persulfate-based AOPs utilize highly-reactive sulfate radical (SO4•–) to degrade organic contaminants [14]. The SO4•– poses a greater redox potential (2.60~3.10 V vs. NHE) and longer half-life time (30~40 µs) than that of •OH (+1.90~+2.70 V vs. NHE and ~20 ns) [15, 16].
The most common persulfates are peroxymonosulfate (PMS) and peroxydisulfate (PDS). Both PMS and PDS have O–O bond, but with different molecular structures (e.g., PMS is asymmetrical while PDS is symmetrical) [17]. Persulfate alone only demonstrated limited oxidative capability that can hardly degrade those organic pollutants, it is necessary to further activate persulfate to generate more aggressive species [18]. PMS and PDS can be activated to generate SO4•– by cleaving their peroxide O−O bond using a range of activation methods such as heat treatment, UV irradiation, and transition metal-based catalysts [14, 19]. However, the large energy inputs required for thermal/UV activation and secondary pollution associated with the use of transition metals limit their practical applications. Thus, heterogeneous catalytic systems based on metal-free carbon nanocatalysts are the preferred strategies [17].
Carbon nanocatalysts have emerged as promising alternatives for persulfate activation. They have widespread availability and low cost, superior biocompatibility, engineered structures, tunable physicochemical properties, and tolerance of harsh environments [20, 21]. Since the discovery of carbon nanomaterials as metal-free persulfate activators for organic compound degradation in 2012 [22], many novel and sustainable carbon nanocatalysts including 0D nanodiamond, 1D carbon nanotubes (CNTs) and carbon nanofibers (CNFs), 2D graphene and graphitic carbon nitride (g-C3N4), and 3D carbon nanocatalysts have been reported for wastewater remediation [20, 23].
To date, there have been some inspiring reviews of metal- and nonmetal-based heterogeneous catalysts for persulfate activated organic compound degradation, while only limited attention has been paid to persulfate activated EOCs remediation. Consequently, a timely review of the progress of metal-free carbon nanocatalysts for persulfate activation was warranted. Here, we present a review of this emerging topic commencing with an introduction to the mechanisms of persulfate activation. The applications of metal-free carbon nanocatalysts for the persulfate activated degradation of EOCs are discussed in detail together with the underlying theory of the removal process. The challenges for improving the practicality of metal-free carbon nanocatalysts and pathways for future research and development in this promising field are highlighted.
2. Mechanisms of persulfate activationPersulfate-based AOPs have been extensively studied for the degradation of pollutants via radical and nonradical pathways. The radical mechanism is based on electron transfer resulting in activation and dissociation of the O−O bond in persulfates to produce SO4•− or •OH or both species for the oxidation of the organic substrates [24]. In the nonradical pathway, the ROS are not typical free radicals. Instead, organic compounds are oxidized or mineralized by other pathways involving direct electron transfer or singlet oxygen (1O2). In processes involving direct electron transfer, carbonaceous material acts as a bridge to facilitate electron transfer from organic pollutants to metastable PDS or PMS without the production of reactive species [25]. 1O2 can be generated by many non-photochemical routes including: (i) The self-decay of peroxy acids such as PMS, peracetic acid, and monoperphthalic acid at pHs exceeds their pKa (~8.2-9.3) [26], (ii) the recombination or direct oxidation of O2•− radical generated catalytically [27], and (iii) the combination of persulfates and a carbonyl moiety on carbonaceous materials [28]. Table 1 compares the reported radical and nonradical pathways of persulfate activation and the different mechanisms of persulfate activation are reviewed below.
|
|
Table 1 A comparison of free radical and non-radical pathway of persulfate activation. |
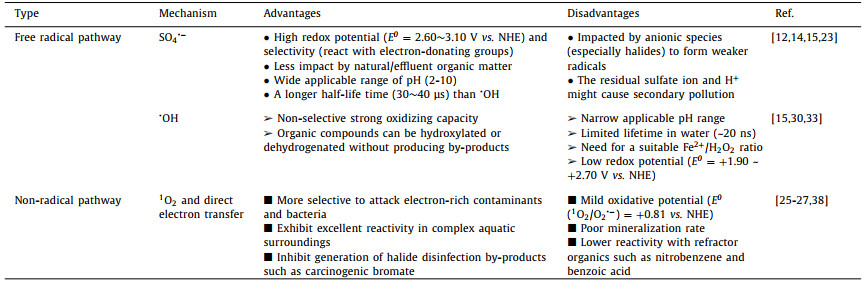
SO4•− has a standard redox potential (E0) of 2.60~3.10 V (vs. NHE) [14, 15]. It has a strong oxidative capability and can be generated in situ by the cleavage of the persulfate molecule peroxide O−O bond via external stimuli and electron transfer reactions. Like •OH, SO4•− can oxidize a range of organic pollutants by three pathways: (i) electron transfer, (ii) addition-elimination, and (iii) hydrogen abstraction [12, 29]. Compared with •OH, SO4•− possess a stronger electron extraction tendency. For example, electrophilic SO4•− selectively attacks aromatic compounds by electron transfer [30, 31]. SO4•− can also rapidly oxidize a range of organic pollutants as well as mineralizing selected compounds. Electron-donating groups on the pollutant such as alkyl, allyl, and hydroxyl (−OH) can increase the reaction rate with SO4•−, while electron-withdrawing groups such as carboxylic (−COOH), carbonyl (C=O), and nitro (−NO2) can decrease the rate of reaction to some extent [24].
2.1.2. •OH-mediated oxidationThe •OH is a well-known powerful oxidant, which can be produced directly from unsymmetrical PMS according to Eq. 1 whereas the production of •OH from symmetrical PDS is dependent on SO4•− and H2O (Eq. 2) [12, 17]. Generally, the organic removal efficiency of •OH is negatively correlated with solution pH [29]. At neutral pH, SO4•− is the dominant reactive radical [32]. Under acidic conditions, SO4•− may also be the dominant reactive radical limiting the generation of H2O2 due to its suppressed reaction with H2O [33]. At pH > 9, the transition from SO4•−-dominated to •OH-dominated oxidation process could readily occur (Eq. 3) [30].
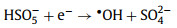
|
(1) |
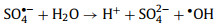
|
(2) |
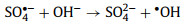
|
(3) |
High-energy 1O2 is a reactive oxidant which has been widely used for photodegradation, organic synthesis, and photodynamic therapy [27, 28]. It reacts almost exclusively with unsaturated organic compounds via electrophilic addition and electron abstraction. Commonly, 1O2 can be formed by the self-decay of PMS at pH above its pKa values (~8.2-9.3; Eq. 4) [34]. Under alkaline conditions, the ketone moieties of organic compounds (e.g., p-benzoquinone and cyclohexanone) have been observed to catalyze 1O2 generation according to the following sequence: nucleophilic addition of PMS to the ketone; formation of a dioxirane intermediate; reaction of the dioxirane with PMS leading to recovery of the ketone and the generation of 1O2 [35]. The carbonyl groups of quinones could also activate PMS to yield 1O2 under basic conditions [36].

|
(4) |

|
(5) |
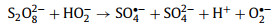
|
(6) |
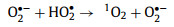
|
(7) |
Recent studies have shown that 1O2 also can be produced by persulfate activation under acidic and neutral conditions [37, 38]. Carbonaceous materials such as CNTs [28], nanodiamond [39], and graphene [40] facilitate 1O2 generation in the presence of persulfate. Carbonyl groups on the surface of these carbonaceous materials were proposed as the active sites for the process. In the case of PDS, an alternative mechanism involving CNTs catalyzed hydrolysis was proposed Eqs. 5 and 6 [12, 41], where O2•− (or HO2•) was regarded as the intermediate to form 1O2 via a proton-promoted disproportionation reaction (Eq. 7) [42].
2.2.2. Direct electron transferApart from the nonradical process via 1O2, the electron transfer pathway involving organic compounds, facilitated by selected activators, could also be responsible for the nonradical activation of persulfate [43, 44]. As reported, transition metals can initiate the one-electron reduction of persulfate to SO4•− (radical mechanism), whereas carbonaceous nanomaterials can mediate electron transfer from organic pollutants (electron donor) to persulfate (electron acceptor) without the generation of radicals [45]. The formation of a reactive persulfate complex on the surface of the highly conductive catalysts may be involved in the electron transfer. Accordingly, N doping of CNTs (N-CNTs) [46] or a graphitic carbon shell formed by the thermal annealing of nanodiamond [47] improved the kinetics of this oxidation scheme due to enhanced surface interaction with the anionic PMS [45]. Similar pathways may occur when other oxyanions such as periodate and peracetate are used with CNTs. The CNTs/oxyanion showed similar substrate-specific reactivity and reaction product formation, regardless of the oxyanion type, which supported the hypothesis that direct electron transfer from organic compounds was likely to be the major oxidation pathway [45].
For example, Ren et al. [48] reported an electron-mediated mechanism for a PDS/CNTs system. They proposed that PDS was initially catalyzed by CNTs to form a complex (CNTs-PDS*) which then selectively abstracted electrons from co-adsorbed phenolic compounds initiating their oxidation without the generation of free radicals. A nonradical pathway was also identified for a PMS/N-CNTs system. Here, the organic compounds were oxidized by N-CNTs-PMS* via an electron-transfer pathway involving the conductive carbon surface [44].
3. Persulfate activation by metal-free carbon nanocatalystsCarbon-based nanomaterials are promising metal-free nanocatalysts due to their desirable attributes associated with cost, availability, activity, and stability [20, 49]. Investigations suggest that the catalytic performance of carbon catalysts is highly dependent on their configuration and dimensional structure [50]. Table 2 summarizes recent developments in carbon allotropes for EOCs removal via persulfate activation based on the following dimensional structures: 0D nanodiamond; 1D CNTs and CNFs; 2D graphene and g-C3N4; and 3D carbon nanocatalysts. The related theory of metal-free carbon nanocatalysts used for the activated persulfate removal of EOCs is discussed below.
|
|
Table 2 Carbon nanocatalysts activation of persulfate for degradation of EOCs. |
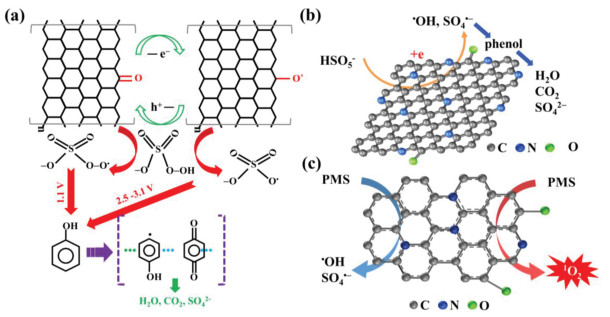
Nanodiamond is a nanomaterial comprising a sp3 diamond core surrounded by a sp2 carbon surface layer resembling the structure of graphite [51]. In 1963, nanodiamond was found to be capable of degrading 2, 4, 6-trinitrotoluene/hexogen in an inert oxygen deficient media without additional carbon sources [52], and other catalytic applications have followed [51]. Duan et al. [53] were the first to report the use of nanodiamond for persulfate activation. The removal efficiency of phenol was 39.5% after 180 min and was much lower than other carbon materials such as single-walled CNTs, mesoporous carbon, and reduced graphine oxide (rGO). Therefore, the surface functionalization of nanodiamond might provide a largely untapped opportunity for its application. As such, annealed nanodiamond was used as the catalyst to activate persulfate for the degradation of phenol [54]. A schematic of the process given in Fig. 1 shows that •OH was the dominant species during the reaction together with minor amounts of SO4•−. Similar observations were also reported for the activation of persulfate using graphene and CNTs [53]. Oxidized water molecules on the surface of the carbocatalyst were essential for the generation of both •OH and SO4•−.

|
Download:
|
| Fig. 1. The mechanism of persulfate activation on annealed 0D nanodiamond. Adapted with permission [54]. Copyright 2016, Elsevier. | |
{kind=link}
Heteroatom doping is an effective method of improving the catalytic performance of nanodiamond. Duan et al. [55] showed that the efficacy of nanodiamond for PMS activation was significantly enhanced after doping with N. They proposed that nucleophilic ketone groups on the nanodiamond shell were the active sites, and the material offered good catalytic potential for active radical generation from PMS via the redox process. Shao et al. [39] studied the mechanism of organic contaminants remediation in nanodiamond-based catalysts by regulating the number of ketone carbonyl groups. Unlike conventional oxidative systems, the dominant reactive oxygen species was identified as 1O2 which exhibited high selectivity in the annealed nanodiamond system enabling the efficient oxidation of 4-chlorophenol. The authors showed that surface ketone (carbonyl) groups were the catalytically active sites, and the rate constant for the oxidation reaction was positively correlated with the amount of these functional groups. Notably, this 1O2-mediated oxidation system outperformed the conventional oxidative processes used by commercial wastewater treatment plants due to its improved selective oxidation capability.
Based on the enhanced catalytic performance resulting from surface functionalization, Lee et al. [56] found that graphited nanodiamond could activate persulfate for phenol removal, and the carbocatalyst outperformed other benchmark materials such as CNTs and rGO. Duan et al. [47] further demonstrated that annealed detonation nanodiamonds could act as efficient bridges for charge transfer due to the unique electronic properties at the sp2 (core) and sp3 (shell) interface which could induce electron transport from core (electron donor) to shell (electron acceptor). Based on density functional theory calculations, the catalytic activity of nanodiamond was promoted by a synergistic effect of charge transport at the interface resulting in an electron-enriched carbon surface. More importantly, an increase in the number of graphite layers on the nanodiamond could change the PMS activation mechanism for catalytic oxidation from a radical-based reaction to a nonradical pathway. Such novel catalytic properties of tunable oxidative potentials via carbocatalysis represents a fascinating prospect for persulfate activated EOCs degradation.
3.2. 1D carbon nanocatalysts 3.2.1. Carbon nanotubesLike graphene, CNTs are composed of sp2 carbon units in hexagonal networks. CNTs exhibit attractive and unique electronic properties that are different from other carbon allotropes due to their unique 1D nanostructure [49, 57]. As metal-free catalysts, CNTs have demonstrated efficient activity for the selective oxidation of ethylbenzene to acetophenone in the liquid phase [58]. Mechanistic studies showed that CNTs played a key role in production of acetophenone involving π-π interactions between the radical species, peroxide, and the graphene sheets.
Moreover, CNTs have been proved as effective metal-free catalysts in the persulfate-based AOPs [59]. Generally, it is recognized that •OH and SO4•− are the main reactive species for organic pollutants degradation by most persulfate-based AOPs. For instance, Sun et al. [59] reported that both PDS and PMS could be activated by pristine CNTs, generating SO4•− for the oxidation of phenol. To enhance catalytic activity of the CNTs, doping was used to incorporate N into the sp2 networks. There was an intrinsic difference between PMS and PDS activation for catalytic oxidation with the N-CNTs. PDS was not effectively activated by the N dopants and could only be initiated by sp2 carbon and oxygen functional groups. It also showed that both CNTs-C=O and CNTs-C=N could transfer an electron to PMS to produce SO4•−, while only CNTs-C=O was effective for PDS activation. Interestingly, the chemical nature of the N dopant had a significant influence on catalytic performance. Pan et al. [60] prepared a series of N-CNTs for the persulfate activated degradation of 2, 4, 4'-trihydroxybenzophenone using urea, NH4NO3, indole, and polyaniline as the N sources. The results showed that the NH4NO3-CNTs−OH catalyst had the best catalytic performance towards 2, 4, 4'-trihydroxybenzophenone removal, due to the combined contribution from •OH and SO4•− in this case.
Both radical and nonradical process were observed using N-doped single-walled CNTs as a metal-free catalyst for PMS activation. It was found that a nonradical pathway was accompanied by the formation of both •OH and SO4•− in the oxidation of phenol with PMS (Fig. 2a) [46]. PMS and phenol were initially adsorbed onto the electron-rich sp2 sites. Activation of PMS then followed (either by radical or nonradical pathways) and subsequent degradation of phenol occurred on the N-CNTs surface. This study provided insights into the role of N heteroatoms in the nanocarbons catalysts.

|
Download:
|
| Fig. 2. Proposed mechanisms for persulfate activation using 1D CNTs: (a) Mechanism of PMS activation on N-Doped CNTs; (b) 1O2 and direct CNTs-mediated electron transfer mechanism for 2, 4-dichlorophenol degradation. (a) is adapted with permission [46]. Copyright 2015, American Chemical Society. (b) is reproduced with permission [64]. Copyright 2019, Elsevier. | |
{kind=link}
Although studies have shown that CNTs could activate persulfate via radical (e.g., •OH and SO4•−) or nonradical pathways, the mechanism of nonradical oxidation by carbocatalysis is still unclear, mainly due to the physicochemical and structural complexity of pure carbon atoms [25]. Besides the commonly reported radical pathways, another nonradical route in the PDS/CNTs system has received increasing attention. Lee et al. [61] used unmodified single and multi-walled CNTs to active persulfate for the degradation of organic compounds. The system showed high reactivity toward phenolic compounds and selected pharmaceuticals (i.e., carbamazepine, propranolol, sulfamethoxazole, and acetaminophen) via a nonradical mechanism. It was suggested that PDS might bind to the surface of the CNTs to form a transient reactive complex responsible for oxidation of the contaminants. However, compounds with electron-withdrawing groups, such as benzoic acid and nitrobenzene (i.e., probes for SO4•− and •OH radicals), were resistant to degradation with this system. The mechanistic reactions could be described a two-stage electron transfer process: (i) formation of reactive complexes with persulfates binding to the surface of the CNTs; and (ii) the subsequent oxidation of phenol by these complexes.
An electron transfer regime was proposed for the nonradical mechanism of persulfate activation on CNTs. Yun et al. [45] demonstrated that CNTs could act as a powerful mediator for the transfer of electrons from 4-chlorophenol (electron donor) to persulfate (electron acceptor) during oxidation. A 100% removal efficiency was achieved in 60 min. The authors also suggested that the inhibitory effect of l-histidine (1O2 scavenger) on the CNTs-activated PMS process was attributable to the rapid depletion of PMS, and the CNTs-mediated electron transfer from pollutants to PMS mainly contributed to the organic contaminant degradation [62]. In a separate report, Ren et al. [63] showed that the improved reactivity of thermally annealed CNTs was due to a consolidated electron-transfer regime without the generation of free radicals and 1O2.
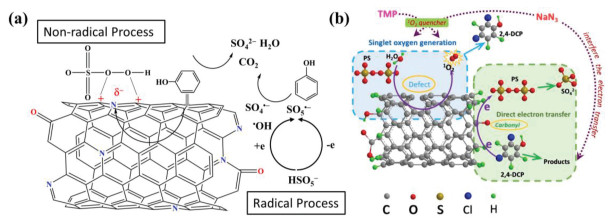
In addition to the direct electron transfer regime, 1O2 degradation of organic pollutants in the CNTs/persulfate system was also shown to involve a nonradical pathway. It is well known that 1O2 is a highly selective oxidant that reacts almost exclusively with electron-rich substances. Cheng et al. [28] reported that 1O2 was capable to oxidizing electron-rich substances in the CNTs/persulfate system and carbonyl groups at the CNTs boundaries were proposed as the active sites for the generation of 1O2. In a subsequent study, Cheng et al. [64] reported that both 1O2 and direct CNTs-mediated electron transfer were involved in persulfate activation by CNTs for the oxidation of 2, 4-dichlorophenol (Fig. 2b). Radical capture, quenching experiments and linear sweep voltammetry measurements confirmed that structural defects and carbonyl groups on the surface of the CNTs played a significant role in the generation of 1O2.
Thermal annealing is a feasible approach to increase the catalytic activity of CNTs by regulating their oxygen content, engineering defects and promoting the degree graphitization. Inspired by this, Yang et al. [42] studied the effects of nitric acid/annealing of CNTs on persulfate activated phenol removal. The results showed that a phenol removal efficiency of 95% could be attained with nitric acid/annealed CNTs compared with a 63% removal efficiency using pristine CNTs. The authors suggested that nitric acid/annealing modified the CNTs resulting in more defective edges and O-containing groups, and subsequent annealing at high temperate facilitated the conversion of sp3 to sp2 carbon. Under these conditions, catalytic activity for persulfate activation was enhanced by the carbonyl group active sites and sp2-hybridized carbon at the defective edges. Furthermore, radical quenching experiments and electron paramagnetic resonance (EPR) spectrometry showed that 1O2 was the main reactive species contributing to phenol removal. This pivotal study may provide a basis for the design of efficient, non-toxic carbocatalysts for the remediation of wastewater.
3.2.2. Carbon nanofibersUnlike CNTs, which typically comprise graphene layers rolled into seamless cylinders with a plane parallel to the longitudinal axis, CNFs can be formed by stacking graphene sheets oriented at various angles (from 0° to 90°) to the fiber axis [65, 66]. CNFs have attracted interest for catalytic applications due to their high aspect ratio, mechanical stability, and high electrical conductivity [67].
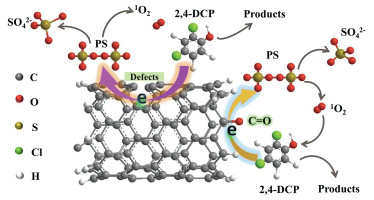
Studies have shown that CNFs can serve as hosts for transition metals nanoparticles such as Co and Ni [68, 69]. However, only limited efforts have been devoted to the applications of CNFs towards persulfate activation and wastewater remediation. In addition, the contribution of the intrinsic properties of CNFs towards persulfate activation performance, like carbon configuration, oxygen-containing functional groups and structural defects, remains controversial. Encouraged by this, Cheng et al. [70] demonstrated a full degradation of 2, 4-dichlorophenol within 60 min, indicating that CNFs were efficient metal-free persulfate activators. The addition of methanol and tert-butyl alcohol to the degradation system had negligible impact on removal efficiencies, suggesting that the oxidative mechanism was not dominated by radicals. However, the addition of sodium azide and l-histidine (1O2 quenching agents) dramatically inhibited removal, confirming the primary role of 1O2 in the degradation of 2, 4-dichlorophenol. Carbonyl groups and structural defects of the surface of the CNFs are active sites for the generation of 1O2 while the sp2 hybridized carbon structure may act as an essential mediator for electron transfer involving the delocalized π electron system (Fig. 3).

|
Download:
|
| Fig. 3. Proposed mechanism for persulfate activation using 1D CNFs. Adapted with permission [70]. Copyright 2020, Elsevier. | |
{kind=link}
Graphene, as the first 2D atomic crystal available, has attracted substantial research interest in water purification due to its desirable physical and chemical properties (e.g., electronic and thermal conductivity, specific surface area (SSA), inherent chemical functionalization, and mechanical strength) [71]. Graphene-based materials such as graphene oxide (GO) and rGO have attracted considerable research interest in the recent decades, particularly in the study of persulfate activation for wastewater remediation [72]. In 2012, Sun et al. [22] were the first to report that rGO with ratios for the intensity of the Raman D and G peaks of > 1.4 (estimation of defect density in graphite) were efficient activators of PMS, yielding SO4•− for the catalytic oxidation of organic pollutants (i.e., phenol, 2, 4-dichlorophenol, and methylene blue). The activity of rGO was shown to be higher than other carbon materials such as activated carbon, graphite powder, GO, and CNTs. It was proposed that surface ketone group on rGO could undergo electron transfer reactions with PMS to produce SO4•−and SO5•− (Fig. 4a). It was also reported that rGO exhibited significant persulfate and PMS activation generating •OH and SO4•− for the degradation of bisphenol A [73]. The proposed heterogeneous activation of persulfate and PMS by rGO and the generation of •OH and SO4•− can be described by Eqs. 8 and 9:
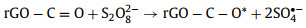
|
(8) |

|
(9) |

|
Download:
|
| Fig. 4. (a) Proposed mechanism of persulfate activation using 2D graphene for phenol degradation. (b) Proposed radical mechanism of PMS activation using N-doped graphene. (c) Proposed nonradical mechanism of PMS activation using N-doped graphene. (a) is adapted with permission [22]. Copyright 2012, American Chemical Society. (b) is adapted with permission [76]. Copyright 2015, American Chemical Society. (c) is adapted with permission [40]. Copyright 2017, The Royal Society of Chemistry. | |
{kind=link}
An important strategy to improve the catalytic performance of graphene for persulfate activation is the introduction of hereroatom dopants such as B, N, S, and P [74]. Sun et al. [75] prepared N-doped rGO (N-rGO) catalysts using a one-pot synthesis and used it for the PMS activated degradation of phenol. The results showed that the catalytic performance of N-rGO was significantly enhanced compared with pure rGO under the same conditions. The degradation efficiencies of phenol (20 mg/L) using N-rGO and rGO after 45 min were 100% and 52.5% respectively. In another study, Duan et al. [76] demonstrated that the catalytic activity of N-rGO was 80 times higher than that of undoped rGO. It was found that both •OH and SO4•− contributed to the oxidation of phenol (Fig. 4b). EPR and radical scavenging experiments subsequently showed that •OH and SO4•− were the main active species in the N-rGO activated PMS degradation of phenol [74]. Liang et al. [40] proposed an alternative nonradical mechanism in the N-rGO/PMS system involving 1O2 as the dominant species for the degradation of phenolic compounds, rather than •OH or SO4•− (Fig. 4c). These findings suggested that the choice of synthetic methods and surface properties of rGO could influence the persulfate catalytic process.
Moreover, Wang et al. [77] used a hydrothermal method to prepare N-rGO and observed crumpled wave-like sheets of graphene in the microstructure. The adsorption and catalysis capabilities of rGO were both enhanced after N modification. An efficient persulfate activated degradation of bisphenols was achieved and attributed to the N modification. This furnished the active sites for persulfate to produce surface bound SO4•− radicals. In a similar study, N-doped graphene was prepared by the thermal annealing of GO and urea and used as a catalyst for the PMS activated degradation of sulfacetamide [78]. The content of reactive functional groups (e.g., graphitic N, pyridinic N, pyrrolic N, nitric oxide, and carbonyl groups) and catalytic performance were carefully controlled by adjusting the annealing temperature. The results showed that N-doped graphene produced at an annealing temperature of 600 ℃ had a relatively high N content (16.0 wt%), and the degradation efficiency for sulfacetamide (10 mg/L) was 100% after 60 min.
Co-doping of graphene using two or three elements with different electronegativities can give unique electron distributions resulting in a synergistic effect. Duan et al. [79] reported that the simultaneous introduction of S and N atoms into graphene sheets could significantly change the surface charge distribution and electrostatic potential of graphene. The S and N co-doped rGO exhibited enhanced PMS activated catalytic oxidation of phenol. In a similar study, N and S doped industrial reduced graphene oxide (i-rGO) were prepared by a thermal decomposition method [80]. The resultant i-rGO-NS was shown to be an effective metal-free catalyst. Degradation experiments showed that methyl paraben was completely removed by i-rGO-NS PMS activation, while i-rGO or i-rGO-N were ineffective activators. These findings could contribute to the development of heteroatom doped industrial graphene-based nanocatalysts, and i-rGO-NS may also have potential as novel metal-free catalyst for environmental pollutants remediation.
To date, most studies have used N-doped graphene or rGO catalysts in powder form and conventional batch reactors for persulfate activation. Notably, the separation of these ultrafine nanoscale powder catalysts post reaction is costly and tedious, limiting the practical application of many promising metal-free catalysts. In addition, mass transfer of target molecules towards the active sites of the catalyst is predominantly controlled by a relatively slow diffusion process, which significantly limits the kinetics of oxidation. Therefore, the development of GO and rGO-based membrane technologies could provide new opportunities to overcome these limitations.
As discussed previously, doping with N heteroatom can enhance the catalytic ability of graphene toward persulfate activation. Inspired by this, Liu et al. [81] exploited the benefits of heteroatom doping by preparing N-rGO filters to activate persulfate for the degradation of phenol. The N-rGO catalyst was synthesized via a hydrothermal method and the N-rGO filters were prepared using a simple vacuum filtration procedure. The rate constant for the oxidation of phenol with the flow-through design was 3.6 times greater than that using a conventional batch system. This was mainly due to the liquid flow through the filter leading to convection-enhanced transfer of the target molecules to the filter active sites, demonstrating the advantage of such a design for practical applications. In light of this, Pedrosa et al. [82] prepared a series of filtration membranes for the degradation of phenol and oxalic acid from GO catalysts synthesized with different precursors (i.e., melamine, urea, and gaseous ammonia) using a modification of the Hummers' method. The melamine modified rGO material had the highest N content (23.78 wt%) and catalytic activity for the degradation of organic compounds in aqueous solution. This superior catalytic performance was ascribed to the presence of N-rich active sites, in particular the N-pyridinic groups. The removal efficiencies of phenol and oxalic acid were 96% and 100%, respectively after 180 min reaction time. 1O2 was determined as the main species responsible for degradation of the target pollutants. More recently, membranes were fabricated using a vacuum filtration method with polyvinylidene fluoride as the support for N-rGO catalysts [83]. The N-rGO membrane was used for the persulfate activated degradation of three fluoroquinolone antibiotics (i.e., ofloxacin, ciprofloxacin, and enrofloxacin) in the continuous flow mode over 24 h. The performance of the composite membrane was demonstrated by complete degradation of the three fluoroquinolone antibiotics present in ultrapure water at 100 µg/L each, and the membrane also exhibited high resistance to fouling. The incorporation of rGO catalysts on a polymeric membrane demonstrates their potential as filters for practical applications in water purification.
3.3.2. Graphitic carbon nitrideg-C3N4 has a stacked structure of one-atom-thick planar sheets and has been considered as a sp2 hybridized N-substituted graphene. It has attracted increasing attention as a promising metal-free catalyst owing to its unique electronic structure, simple synthesis, low cost and toxicity, and high physicochemical stability [84, 85]. g-C3N4 is an established 2D layered polymer semiconductor with a highly delocalized p-conjugated electronic system. Normally, it has a relatively narrow bandgap (~2.7 eV), which endows superior light absorption capacity [85].
The utilization of g-C3N4 in heterogeneous catalysis was first studied in 2006 [86]. Liu et al. [87] subsequently demonstrated the metal-free activation of persulfate by g-C3N4 under visible light irradiation for the decomposition of bisphenol A in aqueous solution as a model reaction. The results indicated that g-C3N4 was capable of activating persulfate to generate superoxide radicals and photogenerated holes, which in turn led to the mineralization of the organic compounds. g-C3N4 also showed good chemical stability and could be used as robust photocatalyst for environmental purification. In another report, the performance of mesoporous g-C3N4 was investigated for the persulfate activated degradation of several organic pollutants including the antibiotics amoxicillin, cefotaxime, meropenem, and sulbactam [88]. The degradation of sulbactam in the mesoporous g-C3N4/persulfate system was pH-dependent and distinctly different from other three antibiotics. This was ascribed to the protonation of persulfate under acidic conditions and subsequent electron transfer between the oxygen atoms of the sulfone group of sulbactam and hydrogen sulfite (HSO3−).
Doping and co-doping metal-free carbon catalysts with different heteroatoms (N, S, C, and O) was shown to be an efficient method to tune the physicochemical properties of g-C3N4. Hence, this strategy permits modification of the band gap structure, extension of the light absorption range, enhanced charge transfer mobility as well as the generation of additional active sites. For example, Gao et al. [89] synthesized O-doped g-C3N4 by a facile thermal polymerization method using urea and oxalic acid dihydrate as the carbocatalyst precursors and oxygen source, respectively. Based on density functional theory calculations and EPR experiments, the electronic structure of g-C3N4 was confirmed to be modulated. The resultant O-doped g-C3N4 exhibited enhanced catalytic activity and stability for the PMS activated degradation of organic pollutants resulting from the formation of electron-poor and electron-rich zones. Compared with pure g-C3N4-based persulfate activation systems, PMS activation by O-doped g-C3N4 involved the formation of 1O2. This resulted from oxidation of PMS over the electron-poor C atoms and the production of •OH and SO4•− due to PMS reduction around the electron-rich O atoms. Under these conditions, 1O2 was the major ROS in the O-doped g-C3N4/PMS system which displayed selective reactivity towards organic contaminants with electron-donating groups. This study provides an opportunity for the development of highly efficient g-C3N4-based catalysts via nonmetal doping for environmental pollutants remediation.
Recently, Zhu et al. [90] demonstrated that the electronic structure of g-C3N4 could be reconfigured by C and O-doping. The results revealed that the doped C and O doping initiated the continuous activation of PMS by electron transfer which enhanced the catalytic degradation of organic pollutants without the requirement for visible light irradiation (Fig. 5). In another study, N-rGO was incorporated into the g-C3N4 matrix by a simple one-step process using melamine and urea as the precursors [91]. The g-C3N4/rGO-N catalyst exhibited improved catalytic performance for PMS activation due to the synergistic effect of the two catalysts. 1O2 was shown to be the dominant reactive component involved in the oxidation of the contaminants. These studies illustrate the value of engineering the surface chemistry to design efficient nanohybrid catalysts for persulfate activation.

|
Download:
|
| Fig. 5. Proposed mechanism of PMS activation using C and N co-doped g-C3N4. Adapted with permission [90]. Copyright 2020, Elsevier. | |
{kind=link}
3D carbon nanomaterials offer many advantages over 0D nanodiamond, 1D nanotubes, and 2D graphene due to their unique morphologies, large specific surface area, well-defined interfaces, and abundant pores [92]. A significant practical benefit of these materials can be attributed to their 3D network which prevents aggregation, ensuring mass transport and sustained performance, offsetting the main disadvantage of other nanoparticles used for environmental applications [93]. Hence, the unique large surface area and continuous pores structure of 3D porous carbon nanomaterials represents an ideal material for the removal of pollutants from contaminated water [94].
Encouraged by the advantages of 3D porous carbon materials, Indrawirawan et al. [50] were the first to use 3D hexagonally-ordered mesoporous carbon as a metal-free catalyst for the PMS activated degradation of phenol solutions (first order rate constant, k = 0.209 min−1). The relatively high degradation rate could be attributed to its large SSA (1129 m2/g) and improved adsorption and reduction abilities compared with other carbon materials such as 0D C60 (SSA = 5 m2/g), 1D single-walled CNTs (SSA = 366 m2/g), and 2D graphene nanoplates (SSA = 103 m2/g). •OH and SO4•– were shown to be responsible for the oxidation of phenol in the PMS/3D hexagonally-ordered mesoporous carbon system. A significant contribution to the degradation of organic pollutants in the PMS/3D carbon system via a nonradical pathway was also suggested. For example, Tang et al. [95] proposed a two-step mechanism for the activation of persulfate by 3D hexagonally-ordered mesoporous carbon. The study highlighted the synergistic effects of radical and nonradical pathways contributing to the decomposition of 2, 4-dichlorophenol. While the nonradical pathway played a key role in the degradation process, the radical pathway was rate determining (Fig. 6a). An electron transfer mediated (on 3D hierarchically ordered porous carbon) direct electron transfer from micro-pollutants (electron donor) to PMS (electron acceptor) was demonstrated for the rapid degradation (< 1 min) of a range of micropollutants (i.e., bisphenol A, sulfamethoxazole, 17α-ethinylestradiol, 4-chlorophenol, 2, 4, 6-trichlorophenol, and asulam) [94]. The ordered porous structure and high surface area of 3D hierarchically ordered porous carbon could maintain the mass transport, and adsorption of the micropollutants and PMS, necessary for the efficient electron transfer from micropollutants to PMS. These studies have made significant contributions to the development of metal-free catalysts for the sustainable remediation of EOCs.

|
Download:
|
| Fig. 6. (a) Persulfate activated degradation of organic compounds on 3D mesoporous carbon. (b) Degradation mechanism of BPA in N-doped hierarchical porous carbon/PMS system (a) is reproduced with permission [95]. Copyright 2018, Elsevier (b) is reproduced with permission [98]. Copyright 2021, Elsevier. | |
{kind=link}
3D carbon aerogels have also aroused interest as metal-free catalysts because of their desirable physicochemical properties (e.g., adsorption, electrical and thermal conductivity, and acid/base stability). Jiang et al. [96] prepared a porous 3D carbon aerogel for the persulfate activated degradation of organic pollutants. Under optimum conditions, a complete removal of rhodamine B was achieved after 60 min of treatment. A ternary mechanism for the oxidative degradation was proposed involving rhodamine B (electron donor), the carbon aerogel (electron mediator) and persulfate (electron acceptor). Interestingly, neither SO4•– nor •OH was generated during persulfate activation and it was suggested that the formation of active complexes on the surface of the carbon aerogel was responsible for rhodamine B degradation. A similar nonradical mechanism was observed in a separate study suggesting that an active complex was initially formed on the catalyst surface which then degraded the target contaminant via electron transfer [97]. Lu et al. [98] also proposed the electron transfer mechanism for the degradation of bisphenol A in a N-doped hierarchical porous carbon aerogels/PMS system. As shown in Fig. 6b, the catalyst acted as a transport channel to facilitate the transfer of electrons from bisphenol A (electron donor) to PMS molecules (electron acceptor) resulting in oxidative degradation of the pollutant. The unique porous/high surface area structures and mass and electron transport capabilities of 3D carbon nanomaterials suggest that these materials will be potential candidates for developments in environmental pollutants remediation.
4. Challenges and perspectivesAlthough studies have demonstrated that metal-free carbon nanocatalysts are promising activators of persulfate, some challenges for their practical application remain, and future research should address these limitations. Firstly, the high cost of engineered the carbon nanomaterials such as CNTs and GO poses a significant challenge for the large-scale production of nanocatalysts. Innovations in carbon nanomaterials are necessary to advance the development of sustainable and low-cost carbon nanomaterials. From an industrial perspective, further efforts are required to manufacture carbon nanomaterials from available raw materials and reduce preparation costs. Consequently, fabrication methods for carbon nanomaterials should be simple, cost-effective, and environmentally friendly.
Secondly, the stability and reusability of carbon nanocatalysts during practical application requires carefully consideration. Most carbon-based nanomaterials in their pure form cannot be used directly for water treatment applications due to their small size, and recovery post remediation is complex and costly. Furthermore, the accumulation of carbon-based nanomaterials in the ecosystem poses potential risks to the receiving ecosystems, which also limits their application. Therefore, the development of carbon nanocatalysts for practical applications should focus the incorporation of these nanomaterials into membranes as well as other methods of recovery [99]. This might entail embedding the nanocatalysts into polymeric beads, or modification with magnetic metal oxides to facilitate their recovery from water. Frequently, these carbon nanomaterials have been embedded in polymer membranes during synthesis or bound to membrane surfaces using physisorption or chemisorption processes. These procedures have allowed the successful development of nanocomposite water treatment membranes.
Inevitably, in-situ environmental applications for pollutants remediation will differ significantly from simplified laboratory scale systems. Many studies measure the oxidation efficiencies of carbon nanocatalysts using pure water containing only target pollutants (thus lacking in background organic content) and often at concentrations much higher than the norm for untreated wastewater. However, natural organic matter such as humic substances from plant decomposition typically exist in much larger quantities than the pollutants of concern, and this is known to decrease persulfate-AOPs efficiencies due to radical scavenging and competitive adsorption onto the carbon nanocatalyst surfaces. Hence, practical applications will require the development of solutions to operate metal-free carbon nanocatalysts under actual wastewater conditions. To achieve sustainable persulfate/carbon nanocatalysts processes, future research should focus on minimizing the occurrence of competing reactions and enhancing the stability of the carbocatalysts during long-term operation, especially in complex water matrices.
5. ConclusionStudies have demonstrated that metal-free carbon nanocatalysts have good potential for the persulfate activated degradation of EOCs in water. In this review, the mechanisms of persulfate activation were discussed in detail including those involving radical and nonradical pathways. Recent advances in metal-free carbon nanocatalysts for the persulfate activated degradation of EOCs were summarized and several strategies (e.g., heteroatom doping, thermal annealing, and filter membrane fabrication) were presented to regulate carbon configurations, surface functional groups and other physicochemical properties. The present challenges and future perspectives toward practical applications were discussed as well.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowldgementsThis work is supported by the Open Project of State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology (No. QAK202108) and the National Natural Science Foundation of China (No. 51822806).
| [1] |
S. Rojas, P. Horcajada, Chem. Rev. 120 (2020) 8378-8415. DOI:10.1021/acs.chemrev.9b00797 |
| [2] |
M.M. Du, Q.Y. Yi, J.H. Ji, et al., Chin. Chem. Lett. 31 (2020) 2803-2808. DOI:10.1016/j.cclet.2020.08.002 |
| [3] |
Y. You, Z.J. Zhao, Y.R. Song, et al., Sep. Purif. Technol. 258 (2021) 117977. DOI:10.1016/j.seppur.2020.117977 |
| [4] |
M. Patel, R. Kumar, K. Kishor, et al., Chem. Rev. 119 (2019) 3510-3673. DOI:10.1021/acs.chemrev.8b00299 |
| [5] |
Y. Xu, T.J. Liu, Y. Zhang, et al., J. Mater. Chem. A 5 (2017) 12001-12014. DOI:10.1039/C7TA03698A |
| [6] |
D.L. Guo, Y.B. Liu, H.D. Ji, et al., Environ. Sci. Technol. 55 (2021) 4045-4053. DOI:10.1021/acs.est.1c00349 |
| [7] |
J. Li, Y.J. Li, Z.K. Xiong, et al., Chin. Chem. Lett. 30 (2019) 2139-2146. DOI:10.1016/j.cclet.2019.04.057 |
| [8] |
B.K. Huang, Z.L. Wu, H.Y. Zhou, et al., J. Hazard. Mater. 412 (2021) 125253. DOI:10.1016/j.jhazmat.2021.125253 |
| [9] |
F.Q. Liu, Y.B. Liu, A.F. Yao, et al., Environ. Sci. Technol. 54 (2020) 5913-5921. DOI:10.1021/acs.est.0c00427 |
| [10] |
R. Anjali, S. Shanthakumar, J. Environ. Manage. 246 (2019) 51-62. DOI:10.1016/j.jenvman.2019.05.090 |
| [11] |
Y.B. Liu, G.D. Gao, C.D. Vecitis, Acc. Chem. Res. 53 (2020) 2892-2902. DOI:10.1021/acs.accounts.0c00544 |
| [12] |
J. Lee, U. von Gunten, J.H. Kim, Environ. Sci. Technol. 54 (2020) 3064-3081. DOI:10.1021/acs.est.9b07082 |
| [13] |
S.L. Nimai, H. Zhang, Z.L. Wu, et al., Chin. Chem. Lett. 31 (2020) 2657-2660. DOI:10.1016/j.cclet.2020.08.008 |
| [14] |
W.D. Oh, Z.L. Dong, T.T. Lim, Appl. Catal. B 194 (2016) 169-201. DOI:10.1016/j.apcatb.2016.04.003 |
| [15] |
P. Neta, R.E. Huie, A.B. Ross, J. Phys. Chem. Ref. Data 17 (1988) 1027-1284. DOI:10.1063/1.555808 |
| [16] |
L.W. Matzek, K.E. Carter, Chemosphere 151 (2016) 178-188. DOI:10.1016/j.chemosphere.2016.02.055 |
| [17] |
S. Waclawek, H.V. Lutze, K. Grubel, et al., Chem. Eng. J. 330 (2017) 44-62. DOI:10.1016/j.cej.2017.07.132 |
| [18] |
I.A. Ike, K.G. Linden, J.D. Orbell, et al., Chem. Eng. J. 338 (2018) 651-669. DOI:10.1016/j.cej.2018.01.034 |
| [19] |
Q.L. Ma, L.C. Nengzi, X.Y. Zhang, et al., Sep. Purif. Technol. 233 (2020) 115978. DOI:10.1016/j.seppur.2019.115978 |
| [20] |
X.G. Duan, H.Q. Sun, S.B. Wang, Acc. Chem. Res. 51 (2018) 678-687. DOI:10.1021/acs.accounts.7b00535 |
| [21] |
Z.Z. Li, C.S. Shen, Y.B. Liu, et al., Appl. Catal. B 260 (2020) 118204. DOI:10.1016/j.apcatb.2019.118204 |
| [22] |
H.Q. Sun, S.Z. Liu, G.L. Zhou, et al., ACS Appl. Mater. Interfaces 4 (2012) 5466-5471. DOI:10.1021/am301372d |
| [23] |
Q.X. Zhao, Q.M. Mao, Y.Y. Zhou, et al., Chemosphere 189 (2017) 224-238. DOI:10.1016/j.chemosphere.2017.09.042 |
| [24] |
U. Ushani, X.Q. Lu, J.H. Wang, et al., Chem. Eng. J. 402 (2020) 126232. DOI:10.1016/j.cej.2020.126232 |
| [25] |
X.G. Duan, H.Q. Sun, Z.P. Shao, et al., Appl. Catal. B 224 (2018) 973-982. DOI:10.1016/j.apcatb.2017.11.051 |
| [26] |
A.D. Bokare, W. Choi, Environ. Sci. Technol. 49 (2015) 14392-14400. DOI:10.1021/acs.est.5b04119 |
| [27] |
Q.Y. Yi, J.H. Ji, B. Shen, et al., Environ. Sci. Technol. 53 (2019) 9725-9733. DOI:10.1021/acs.est.9b01676 |
| [28] |
X. Cheng, H.G. Guo, Y.L. Zhang, et al., Water Res. 113 (2017) 80-88. DOI:10.1016/j.watres.2017.02.016 |
| [29] |
J.F. Yu, H.P. Feng, L. Tang, et al., Prog. Mater. Sci. 111 (2020) 100654. DOI:10.1016/j.pmatsci.2020.100654 |
| [30] |
J.H. Wang, S.Z. Wang, Chem. Eng. J. 334 (2018) 1502-1517. DOI:10.1016/j.cej.2017.11.059 |
| [31] |
C.T. Guan, J. Jiang, S.Y. Pang, et al., Water Res. 176 (2020) 115725. DOI:10.1016/j.watres.2020.115725 |
| [32] |
I.M. Kolthoff, I.K. Miller, J. Am. Chem. Soc. 73 (1951) 3055-3059. DOI:10.1021/ja01151a024 |
| [33] |
C.J. Liang, H.W. Su, Ind. Eng. Chem. Res. 48 (2009) 5558-5562. DOI:10.1021/ie9002848 |
| [34] |
F.E. Dennis, W.U. Mark, J. Chem. Soc., Dalton Trans. (1985) 1151-1153.
|
| [35] |
A. Lange, H.D. Brauer, J. Chem. Soc., Perkin Trans. 2 (1996) 805-811.
|
| [36] |
Y. Zhou, J. Jiang, Y. Gao, et al., Environ. Sci. Technol. 49 (2015) 12941-12950. DOI:10.1021/acs.est.5b03595 |
| [37] |
Y.W. Gao, Y. Zhu, Z.H. Chen, et al., Chem. Eng. J. 394 (2020) 123936. DOI:10.1016/j.cej.2019.123936 |
| [38] |
R. Luo, M.Q. Li, C.H. Wang, et al., Water Res. 148 (2019) 416-424. DOI:10.1016/j.watres.2018.10.087 |
| [39] |
P.H. Shao, J.Y. Tian, F. Yang, et al., Adv. Funct. Mater. 28 (2018) 1705295. DOI:10.1002/adfm.201705295 |
| [40] |
P. Liang, C. Zhang, X.G. Duan, et al., Environ. Sci. Nano 4 (2017) 315-324. DOI:10.1039/C6EN00633G |
| [41] |
O.S. Furman, A.L. Teel, R.J. Watts, Environ. Sci. Technol. 44 (2010) 6423-6428. DOI:10.1021/es1013714 |
| [42] |
W.C. Yang, Z. Jiang, X.X. Hu, et al., Chemosphere 220 (2019) 514-522. DOI:10.1016/j.chemosphere.2018.12.136 |
| [43] |
P.H. Shao, S.P. Yu, X.G. Duan, et al., Environ. Sci. Technol. 54 (2020) 8464-8472. DOI:10.1021/acs.est.0c02645 |
| [44] |
W. Ren, G. Nie, P. Zhou, et al., Environ. Sci. Technol. 54 (2020) 6438-6447. DOI:10.1021/acs.est.0c01161 |
| [45] |
E.T. Yun, H.Y. Yoo, H. Bae, et al., Environ. Sci. Technol. 51 (2017) 10090-10099. DOI:10.1021/acs.est.7b02519 |
| [46] |
X.G. Duan, H.Q. Sun, Y.X. Wang, et al., ACS Catal. 5 (2015) 553-559. DOI:10.1021/cs5017613 |
| [47] |
X.G. Duan, Z.M. Ao, H.Y. Zhang, et al., Appl. Catal. B 222 (2018) 176-181. DOI:10.1016/j.apcatb.2017.10.007 |
| [48] |
W. Ren, L.L. Xiong, X.H. Yuan, et al., Environ. Sci. Technol. 53 (2019) 14595-14603. DOI:10.1021/acs.est.9b05475 |
| [49] |
J.L. Peng, Y.L. He, C.Y. Zhou, et al., Chin. Chem. Lett. 32 (2021) 1626-1636. DOI:10.1016/j.cclet.2020.10.026 |
| [50] |
S. Indrawirawan, H.Q. Sun, X.G. Duan, et al., Appl. Catal. B 179 (2015) 352-362. DOI:10.1016/j.apcatb.2015.05.049 |
| [51] |
A. Krueger, Chem. Eur. J. 14 (2008) 1382-1390. DOI:10.1002/chem.200700987 |
| [52] |
V.V. Danilenko, Phys. Solid State 46 (2004) 595-599. DOI:10.1134/1.1711431 |
| [53] |
X.G. Duan, H.Q. Sun, J. Kang, et al., ACS Catal. 5 (2015) 4629-4636. DOI:10.1021/acscatal.5b00774 |
| [54] |
X.G. Duan, C. Su, L. Zhou, et al., Appl. Catal. B 194 (2016) 7-15. DOI:10.1016/j.apcatb.2016.04.043 |
| [55] |
X.G. Duan, Z.M. Ao, D.G. Li, et al., Carbon 103 (2016) 404-411. DOI:10.1016/j.carbon.2016.03.034 |
| [56] |
H.S. Lee, H.I. Kim, S. Weon, et al., Environ. Sci. Technol. 50 (2016) 10134-10142. DOI:10.1021/acs.est.6b02079 |
| [57] |
M.F.L. De Volder, S.H. Tawfick, R.H. Baughman, et al., Science 339 (2013) 535-539. DOI:10.1126/science.1222453 |
| [58] |
J. Luo, F. Peng, H. Yu, et al., ChemCatChem 5 (2013) 1578-1586. DOI:10.1002/cctc.201200603 |
| [59] |
H.Q. Sun, C.K. Kwan, A. Suvorova, et al., Appl. Catal. B 154 (2014) 134-141. |
| [60] |
X.Y. Pan, J. Chen, N.N. Wu, et al., Water Res. 143 (2018) 176-187. DOI:10.1016/j.watres.2018.06.038 |
| [61] |
H.S. Lee, H.J. Lee, J. Jeong, et al., Chem. Eng. J. 266 (2015) 28-33. DOI:10.1016/j.cej.2014.12.065 |
| [62] |
E.T. Yun, J.H. Lee, J. Kim, et al., Environ. Sci. Technol. 52 (2018) 7032-7042. DOI:10.1021/acs.est.8b00959 |
| [63] |
W. Ren, L.L. Xiong, G. Nie, et al., Environ. Sci. Technol. 54 (2019) 1267-1275. |
| [64] |
X. Cheng, H.G. Guo, Y.L. Zhang, et al., Water Res. 157 (2019) 406-414. DOI:10.1016/j.watres.2019.03.096 |
| [65] |
R. Gusain, N. Kumar, S.S. Ray, Coordin. Chem. Rev. 405 (2020) 213111. |
| [66] |
B. Zhang, F.Y. Kang, J.M. Tarascon, et al., Prog. Mater. Sci. 76 (2016) 319-380. DOI:10.1016/j.pmatsci.2015.08.002 |
| [67] |
D.S. Yu, E. Nagelli, F. Du, et al., J. Phys. Chem. Lett. 1 (2010) 2165-2173. DOI:10.1021/jz100533t |
| [68] |
Y.P. Bao, M. Tian, S.K. Lua, et al., Chemosphere 245 (2020) 125407. DOI:10.1016/j.chemosphere.2019.125407 |
| [69] |
Y.J. Yao, J. Zhang, M.X. Gao, et al., J. Colloid Interface Sci. 529 (2018) 100-110. DOI:10.1016/j.jcis.2018.05.077 |
| [70] |
X. Cheng, H.G. Guo, W. Li, et al., Chem. Eng. J. 396 (2020) 125107. DOI:10.1016/j.cej.2020.125107 |
| [71] |
H. Chen, K.C. Carroll, Environ. Pollut. 215 (2016) 96-102. DOI:10.1016/j.envpol.2016.04.088 |
| [72] |
K.J. Yang, J. Wang, X.X. Chen, et al., Environ. Sci. Nano 5 (2018) 1264-1297. DOI:10.1039/C8EN00194D |
| [73] |
T. Olmez-Hanci, I. Arslan-Alaton, S. Gurmen, et al., J. Hazard. Mater. 360 (2018) 141-149. DOI:10.1016/j.jhazmat.2018.07.098 |
| [74] |
C. Wang, J. Kang, H.Q. Sun, et al., Carbon 102 (2016) 279-287. DOI:10.1016/j.carbon.2016.02.048 |
| [75] |
H.Q. Sun, Y.X. Wang, S.Z. Liu, et al., Chem. Commun. 49 (2013) 9914-9916. DOI:10.1039/c3cc43401j |
| [76] |
X.G. Duan, Z.M. Ao, H.Q. Sun, et al., ACS Appl. Mater. Interfaces 7 (2015) 4169-4178. DOI:10.1021/am508416n |
| [77] |
X.B. Wang, Y.L. Qin, L.H. Zhu, et al., Environ. Sci. Technol. 49 (2015) 6855-6864. DOI:10.1021/acs.est.5b01059 |
| [78] |
X. Chen, W.D. Oh, Z.T. Hu, et al., Appl. Catal. B 225 (2018) 243-257. DOI:10.1016/j.apcatb.2017.11.071 |
| [79] |
X.G. Duan, K. O'Donnell, H.Q. Sun, et al., Small 11 (2015) 3036-3044. DOI:10.1002/smll.201403715 |
| [80] |
P. Sun, H. Liu, M.B. Feng, et al., Appl. Catal. B 251 (2019) 335-345. DOI:10.1016/j.apcatb.2019.03.085 |
| [81] |
Y.B. Liu, L. Yu, C.N. Ong, et al., Nano Res. 9 (2016) 1983-1993. DOI:10.1007/s12274-016-1089-7 |
| [82] |
M. Pedrosa, G. Drazic, P.B. Tavares, et al., Chem. Eng. J. 369 (2019) 223-232. DOI:10.1016/j.cej.2019.02.211 |
| [83] |
O. Vieira, R.S. Ribeiro, M. Pedrosa, et al., Chem. Eng. J. 402 (2020) 126117. DOI:10.1016/j.cej.2020.126117 |
| [84] |
W.J. Ong, L.L. Tan, Y.H. Ng, et al., Chem. Rev. 116 (2016) 7159-7329. DOI:10.1021/acs.chemrev.6b00075 |
| [85] |
Y. Yang, X. Li, C.Y. Zhou, et al., Water Res. 184 (2020) 116200. DOI:10.1016/j.watres.2020.116200 |
| [86] |
F. Goettmann, A. Fischer, M. Antonietti, et al., Chem. Commun. (2006) 4530-4532. |
| [87] |
B.C. Liu, M. Qiao, Y.B. Wang, et al., Chemosphere 189 (2017) 115-122. DOI:10.1016/j.chemosphere.2017.08.169 |
| [88] |
M.M. Dou, J. Wang, B.R. Gao, et al., Chem. Eng. J. 394 (2020) 124899. DOI:10.1016/j.cej.2020.124899 |
| [89] |
Y.W. Gao, Y. Zhu, L. Lyu, et al., Environ. Sci. Technol. 52 (2018) 14371-14380. DOI:10.1021/acs.est.8b05246 |
| [90] |
Y. Zhu, Z.H. Chen, Y.W. Gao, et al., J. Hazard. Mater. 394 (2020) 122578. DOI:10.1016/j.jhazmat.2020.122578 |
| [91] |
P. Sun, H. Liu, M.B. Feng, et al., Appl. Catal. B 272 (2020) 119005. DOI:10.1016/j.apcatb.2020.119005 |
| [92] |
A.B. Jorge, R. Jervis, A.P. Periasamy, et al., Adv. Energy Mater. 10 (2019) 1902494. |
| [93] |
Y. Shen, Q.L. Fang, B.L. Chen, Environ. Sci. Technol. 49 (2015) 67-84. DOI:10.1021/es504421y |
| [94] |
C.H. Chu, J. Yang, D.H. Huang, et al., Environ. Sci. Technol. 53 (2019) 10352-10360. DOI:10.1021/acs.est.9b03067 |
| [95] |
L. Tang, Y.N. Liu, J.J. Wang, et al., Appl. Catal. B 231 (2018) 1-10. DOI:10.1016/j.apcatb.2018.02.059 |
| [96] |
L.L. Jiang, Y. Zhang, M.H. Zhou, et al., J. Hazard. Mater. 358 (2018) 53-61. DOI:10.1016/j.jhazmat.2018.06.048 |
| [97] |
L.L. Jiang, Q. Wang, M.H. Zhou, et al., Chemosphere 241 (2020) 125066. DOI:10.1016/j.chemosphere.2019.125066 |
| [98] |
K.R. Lu, Z.J. Min, J.X. Qin, et al., Sci. Total Environ. 752 (2021) 142282. DOI:10.1016/j.scitotenv.2020.142282 |
| [99] |
Y.B. Liu, F.Q. Liu, N. Ding, et al., Chin. Chem. Lett. 31 (2020) 2539-2548. |