 2021, Vol. 32
2021, Vol. 32 


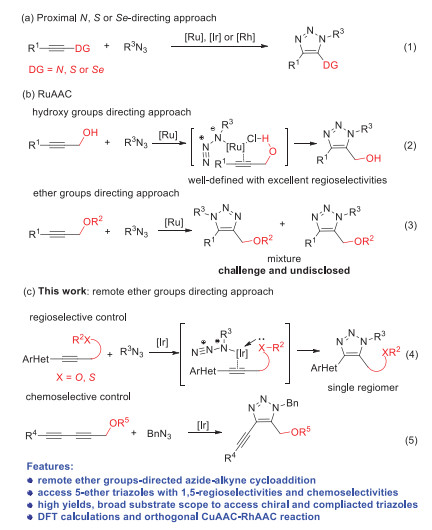
Coordination of directing groups to metals usually play important roles as driving force and control manner for various transformations such as asymmetric hydrogenation, epoxidation and C-H activation [1-8]. However, the application of such coordination strategy is severely restricted by the following two key issues. The nitrogen-, sulfur- or selenium-chelating groups owning strong coordination abilities are more commonly used as directing groups rather than oxygen, especially for ethers with poor coordination to late transition metals, which lead to very limited methodologies for the ether-directed transformations [4]. In addition, most of the current coordination strategies are based on proximal directing groups due to spatial and geometrical accessibility. It is still distinct and longstanding challenge for controlling the selectivity by remote directing groups [9-12]. Albeit some promising progress have been accomplished in overcoming above-mentioned issues in C-H activation [1-12], achieving regioselective and chemoselective cycloaddition of azides and inert alkynes remains undisclosed, especially when the cycloaddition site is far away from directing groups. Herein, we set out to address above problems by applying the weak and remote coordination of ethers to the metals for highly regioselective and chemoselective cycloaddition of azides and alkynes.
Metal-catalyzed azide–alkyne cycloaddition (MAAC) has been well-defined during the past decades to afford various 1,2,3-triazoles [13-16]. However, most of the reactions are limited to use terminal alkynes because of low reactivities and complicated selectivities for electron-rich internal alkynes. Until recently, Mascareñas, López, Sun, Jia, Hong, Cui, our group and others have successfully synthesized a series of 1,4,5-fully substituted 1,2,3-triazoles starting from internal alkynes with excellent regioselectivities using nitrogen-, sulfur- or seleno groups as proximal and strong directing groups to coordinate with various transition metals including Ru, Ir, Rh and Ni (Scheme 1a1) [17-34]. Lin, Jia and Fokin's group used the hydroxyl group from internal propargyl alcohol as directing group to accomplish 1,5-regioselectivities by ruthenium-catalyzed azide–alkyne cycloaddition (RuAAC) [35]. The hydroxyl group as hydrogen bond donor could coordinate with ligating chloride from ruthenium catalyst to favor the 1,5-regioselectivities (Scheme 1b2). Unfortunately, such RuAAC reaction lacks both the regioselectivities and chemoselectivities if the directing group was replaced by ether group, which was mainly due to the weak coordination ability for ether as hydrogen bond acceptor (Scheme 1b3) [36-37]. Moreover, remote directing group has never been applied in tuning selectivity in cycloaddition, which also significantly limited the further application of MAAC reactions [38-42]. As known that ether is one of the most fundamental groups in natural products, drug molecules, polymers and materials [43]. Numerous compounds with diverse biological activities also contain the ether-substituted triazoles [44-46]. Therefore, it is of great significance and interest to develop a direct approach to access fully substituted 5-ether-1,2,3-triazoles with high regioselectivities and chemoselectivities by remote ether groups-directed cycloaddition reaction. Considering that the failure for ether-directed AAC reactions, and as a continuation of our pursuit of highly regioselective cycloaddition reaction, we hypothesized that iridium(I) with stronger coordination ability to the lone-pair electron of ether groups could potentially solve the remote-controlled issues. Thus, we used ether group as directing group to coordinate with iridium catalyst, which resulted in excellent 1,5-regioselectivities and excellent chemoselectivities for the synthesis of various fully substituted 5-ether-1,2,3-triazoles. Beyond that, we found that even the remote ether group (up to four σ bonds away from internal alkyne part) can also control the regioselectivities well during the IrAAC process (Scheme 1c).

|
Download:
|
| Scheme 1. Coordination strategy for regioselective cycloaddition of azides and alkynes. | |
{kind=link}
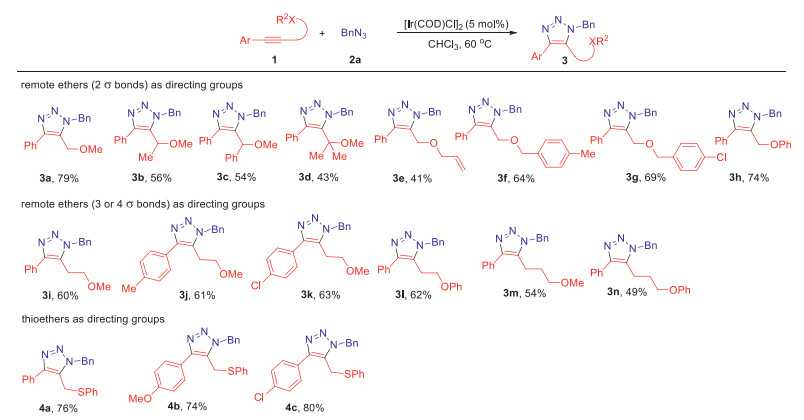
With the optimized conditions in hand (For the details, please see Supporting information), we next examined various substrates with different directing groups in Scheme 2. Various internal alkynes (1), and benzyl azide (2a) were used as substrates to afford fully substituted 5-ether-1,2,3-triazoles (3) in good yields (up to 80%), and excellent regioselectivities (more than 20:1). The steric hindrance effect for ether directing groups was firstly investigated (3a-3d). The yield would dramatically decrease when the steric hindrance was increased from proton (3a) to gem-dimethyl group (3d) albeit with excellent 1,5-regioselectivities. Remarkably, the secondary propargyl ethers could be used as directing groups for IrAAC reactions (3b and 3c), which paved the route to access chiral triazoles from chiral propargyl ethers later. When allyl ether was used as directing group, the yield dropped significantly (3e), which may be attributed to the side reactions involving Ir-π-allyl intermediate [47, 48]. If benzyl ethers were used instead of methyl ether as directing groups, the moderate yields were obtained and no obvious electronic effect was observed (3f and 3g). Aryl ether could also be used as directing group for this transformation with slightly lower yield (3h) compared to methyl ether. Subsequently, the long-distance ether directing groups were explored by extending the σ bonds away from internal alkyne part. Encouragingly, the remote directing groups with three σ bonds away from the alkyne moieties remained well controlling the regioselectivities for IrAAC reaction (more than 20:1), despite that the yields would decrease (3i-3l). Only moderate yields were acquired using remote ethers with four σ bonds away from the alkynes (3m and 3n). Besides various ethers, although the internal thioalkynes have already been successfully used in MAAC process before [18, 19, 25-31], we further explored other kinds of alkynes with thio-functional moieties using thioethers as directing groups for this transformation. To our delight, good yields and regioselectivities were acquired using thioethers as directing groups for various internal alkynes without significant electronic effect (4a-4c).

|
Download:
|
| Scheme 2. Substrate scope using various directing groups. Standard reaction conditions: 1 (1.0 equiv.), 2 (1.5 equiv.), [Ir(COD)Cl]2 (5 mol%), CHCl3 (0.1 mol/L), 60 ℃ for 12–24 h. Yield of isolated product. The regioisomeric ratios of all examples were more than 20:1, which were determined by 1H NMR of the crude mixture with an internal standard. | |
{kind=link}
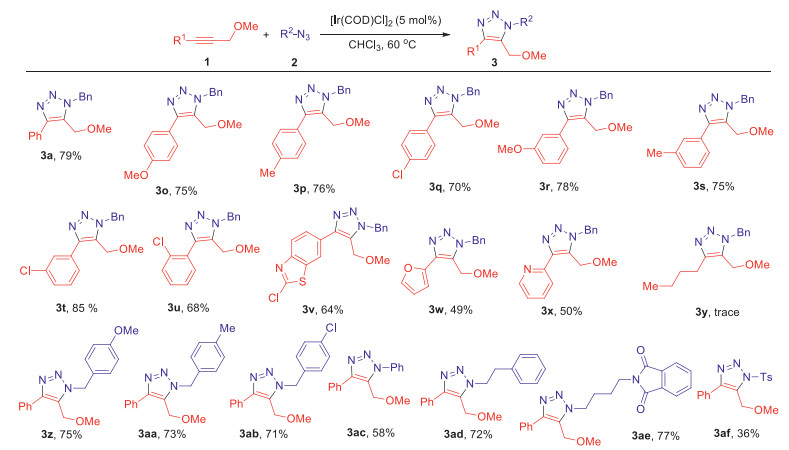
Methyl ether has already been demonstrated to be the most efficient directing group in the IrAAC process. Thus, we continuously explored the substrate ranges of internal alkynes (1) and azides (2) using methyl ethers as directing groups in Scheme 3. Both electron-donating and electron-withdrawing internal alkynes could participate in the cycloadditions with benzyl azide to afford fully substituted 5-ether-1,2,3-triazoles with similar yields (3a, 3o-3t). The electronic effect was not very obvious for para- and meta-methyl, methoxy, chloro substituted internal alkynes (3o-3t). But for the ortho-chloro substituted internal alkyne, the yield would drop to 68% due to the steric hindrance (3u). Noticeably, heterocyclic substrates such as benzothiazole, furan and pyridine could be tolerated in this transformation despite the moderate yields (3v-3x). In these cases, ether group could well control the regioselectivites in a competitive manner with the heteroatoms existing in furan and pyridine, whose coordination abilities may be weakened by aromaticity effect. Unfortunately, only trace amount of product was observed using alkyl internal alkyne as substrate (3y). Then the substrate scope of different azides was screened. The IrAAC reaction could conduct smoothly with various alkyl and aryl azides (3z, 3aa-3ae). Both the electron-donating and electron-withdrawing azides could provide the corresponding products with similar yields (71%−75%), hinting the electronic effect was not obvious for azides (3z, 3aa and 3ab). However, lower yield (3ac) was obtained using phenyl azide instead of alkyl azide. To our delight, ethyl and n-butyl azides could be used as substrates to give complicated fully substituted 5-ether-1,2,3-triazoles with good yields (72% and 77%) (3ad and 3ae). The TsN3 could also participate in this transformation albeit the low yield (3af).

|
Download:
|
| Scheme 3. Substrate scope for methyl ether-directing regioselective cycloaddition. The reactions were set up in standard reaction conditions. | |
{kind=link}
After achieving remote ether groups-directed regioselective cycloaddition of azides and alkynes, we turned to investigate the chemoselective issues by ether groups-directed strategy (Scheme 4). 1,3-Diynyl ethers (5) were designed as substrates for the chemoselective synthesis of fully substituted 4-alkynyl-triazoles (6) by discriminating two different internal alkynes attaching with/without ether groups. The IrAAC reaction only occurred at the propargly ether moiety rather than outer internal alkyne to generate fully substituted 4-alkynyl-triazole, and no any bis-1,2,3-triazole was isolated. Besides methyl-ether could be used as directing group (6a), other ether groups including benzyl-ether and bulky-ether could also promote the transformation well with excellent regioselectivities and chemoselectivities (6b and 6c). For para-substituted diynes, both electron-donating and electron-withdrawing diynes could supply fully substituted 4-alkynyl-triazoles (6d-6f) with similar yields, even for using strong electron-withdrawing nitro substrate (6f). The cycloaddition reaction could proceed smoothly for meta- and ortho-substituted diynes (6g-6i) in spite of lower yield for ortho-chloro substituted diyne (6i). Encouragingly, the diyne with thiophene structure could also be employed as substrate to give 4-alkynyl-triazole (6j) in good yield, and excellent regioselectivity and chemoselectivity. The moderate yields were attributed to the low conversion of internal alkynes.

|
Download:
|
| Scheme 4. Substrate scope for chemoselective synthesis of fully substituted 4-alkynyl-triazoles. The reactions were set up in standard reaction conditions. | |
{kind=link}
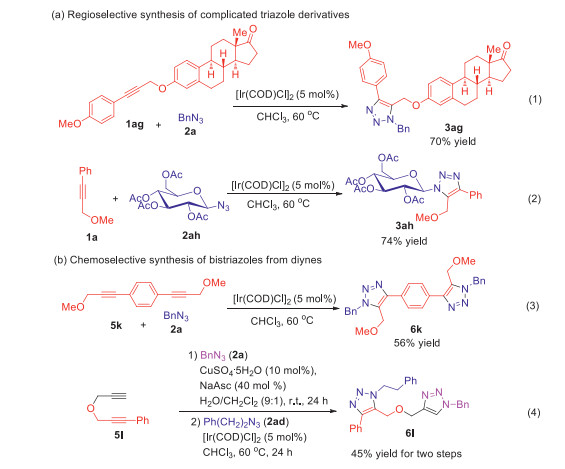
Subsequently, the applicability of ether-directing IrAAC reaction was investigated in Scheme 5. The regioisomeric ratios of all examples in Scheme 5 were more than 20:1, which were determined by 1H NMR of the crude mixture with an internal standard. Estrone as a steroidal estrogen could be efficiently modified by ether-directing IrAAC reaction to form derivate 3ag, which provides a novel drug conjugate approach (Scheme 1a1). Glycosyl azide (2ah) could be utilized to prepare carbohydrate derivate 3ah, which offers the unique and efficient way to access non-natural glycosyl triazoles in glycomics study (Scheme 5a2). The chemoselective synthesis of bistriazoles from other types of diynes was expanded (Scheme 2b). Using symmetric dipropargyl methyl ether (5k) as substrate, fully substituted 5-ether-bistriazole (6k) was acquired with excellent regioselectivity, which could be applied in regiospecific polymerization in future (Scheme 5b3). Terminal alkyne and internal alkyne could be successfully differentiated by orthogonal CuAAC-IrAAC reaction with excellent chemoselectivity. Benzyl azide (2a) and phenylethyl azide (2ad) were used in CuAAC and IrAAC respectively. 1, 6-Diyne (5i) was converted to bistriazole (6l) without cross-reactivity, which could be sequentially modified by mutually orthogonal MAAC approach to rapidly build multi-functionalized molecular scaffolds (Scheme 5b4).

|
Download:
|
| Scheme 5. The application of the regio- and chemoselectively ether groups-directing cycloaddition. | |
{kind=link}
In summary, we have used ether group as directing group to coordinate with iridium catalyst in the cycloaddition process, which results in the excellent 1,5-regioselectivities and chemoselectivities for the synthesis of fully substituted 5-ether-1,2,3-triazoles. We have also demonstrated the remote ether group (up to four σ bonds away from alkyne) could well control the regioselectivities. The IrAAC reaction chemoselectively occurs at the propargyl ether moiety of diynes to give unique fully substituted 4-alkynyl-triazoles. Further applications of this ether groups-directed IrAAC reaction in materials science, medicinal chemistry and polymer chemistry are underway in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by grants from the National Natural Science Foundation of China (No. 21978039), Special Funds of the Central Government Leading Local Government for the Technology Development (Nos. 2021JH6/10500146, 2021JH6/10500148), the Fundamental Research Funds for the Central Universities (Nos. DUT20YG120, DUT19LK60).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.05.037.
| [1] |
R.L. Reyes, M. Sato, T. Iwai, et al., Science 369 (2020) 970-974. DOI:10.1126/science.abc8320 |
| [2] |
R.A. Johnson, K.B. Sharpless, Catalytic Asymmetric Synthesis. 2nd ed.. New York: Wiley, 2005.
|
| [3] |
Z. Li, H. Yamamoto, Acc. Chem. Res. 46 (2013) 506-508. DOI:10.1021/ar300216r |
| [4] |
G. Li, D. Leow, L. Wan, J.Q. Yu, Angew. Chem. Int. Ed. 52 (2013) 1245-1247. DOI:10.1002/anie.201207770 |
| [5] |
Á. Iglesias, R. Álvarez, Á.R. de Lera, K. Muñiz, Angew. Chem. Int. Ed. 51 (2012) 2225-2228. DOI:10.1002/anie.201108351 |
| [6] |
C.W. Liskey, J.F. Hartwig, J. Am. Chem. Soc. 134 (2012) 12422-12425. DOI:10.1021/ja305596v |
| [7] |
J. Oyamada, M. Nishiura, Z. Hou, Angew. Chem. Int. Ed. 50 (2011) 10720-10723. DOI:10.1002/anie.201105636 |
| [8] |
S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka, M. Sawamura, J. Am. Chem. Soc. 131 (2009) 5058-5059. DOI:10.1021/ja9008419 |
| [9] |
G.J. Choi, Q. Zhu, D.C. Miller, C.J. Gu, R.R. Knowles, Nature 539 (2016) 268-271. DOI:10.1038/nature19811 |
| [10] |
X.C. Wang, W. Gong, L.Z. Fang, et al., Nature 519 (2015) 334-338. DOI:10.1038/nature14214 |
| [11] |
D. Leow, G. Li, T.S. Mei, J.Q. Yu, Nature 486 (2012) 518-522. DOI:10.1038/nature11158 |
| [12] |
G. He, G.A. Chen, Angew. Chem. Int. Ed. 50 (2011) 5192-5196. DOI:10.1002/anie.201100984 |
| [13] |
H.C. Kolb, M.G. Finn, K.B. Sharpless, Angew. Chem. Int. Ed. 40 (2001) 2004-2021. DOI:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5 |
| [14] |
C.W. Tornøe, C. Christensen, M. Meldal, J. Org. Chem. 67 (2002) 3057-3064. DOI:10.1021/jo011148j |
| [15] |
V.V. Rostovtssev, L.G. Green, V.V. Fokin, K.B. Sharpless, Angew. Chem. Int. Ed. 41 (2002) 2596-2599. DOI:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| [16] |
B.T. Worrell, J.A. Malik, V.V. Fokin, Science 340 (2013) 457-460. DOI:10.1126/science.1229506 |
| [17] |
J. Miguel-Ávila, M. Tomás-Gamasa, A.P. Olmos, J. Pérez, J.L. Mascareñas, Chem. Sci. 9 (2018) 1947-1952. DOI:10.1039/C7SC04643J |
| [18] |
P. Destito, J.R. Couceiro, H. Faustino, F. López, J.L. Mascareñas, Angew. Chem. Int. Ed. 56 (2017) 10766-10770. DOI:10.1002/anie.201705006 |
| [19] |
S. Ding, G. Jia, J. Sun, Angew. Chem. Int. Ed. 53 (2014) 1877-1880. DOI:10.1002/anie.201309855 |
| [20] |
Q. Luo, G. Jia, J. Sun, Z. Lin, J. Org. Chem. 79 (2014) 11970-11980. DOI:10.1021/jo5018348 |
| [21] |
W.G. Kim, M.E. Kang, J.B. Lee, et al., J. Am. Chem. Soc. 139 (2017) 12121-12124. DOI:10.1021/jacs.7b06338 |
| [22] |
W.G. Kim, S.Y. Baek, S.Y. Jeong, et al., Org. Biomol. Chem. 18 (2020) 3374-3381. DOI:10.1039/D0OB00579G |
| [23] |
L. Zeng, Z. Lai, C. Zhang, H. Xie, S. Cui, Org. Lett. 22 (2020) 2220-2224. DOI:10.1021/acs.orglett.0c00394 |
| [24] |
R. Chen, L. Zeng, Z. Lai, S. Cui, Adv. Synth. Catal. 361 (2019) 989-994. DOI:10.1002/adsc.201801410 |
| [25] |
M. Li, N. Zheng, J. Li, Y. Zheng, W. Song, Green Chem 22 (2020) 2394-2398. DOI:10.1039/D0GC00555J |
| [26] |
W. Song, N. Zheng, M. Li, et al., Org. Lett. 20 (2018) 6705-6709. DOI:10.1021/acs.orglett.8b02794 |
| [27] |
W. Song, N. Zheng, Org. Lett. 19 (2017) 6200-6203. DOI:10.1021/acs.orglett.7b03123 |
| [28] |
W. Song, M. Li, K. Dong, Y. Zheng, Adv. Synth. Catal. 361 (2019) 5258-5263. DOI:10.1002/adsc.201901014 |
| [29] |
W. Song, N. Zheng, M. Li, et al., Adv. Synth. Catal. 361 (2019) 469-475. DOI:10.1002/adsc.201801216 |
| [30] |
W. Song, N. Zheng, M. Li, K. Ullah, Y. Zheng, Adv. Synth. Catal. 360 (2018) 2429-2434. DOI:10.1002/adsc.201800336 |
| [31] |
M. Li, K. Dong, Y. Zheng, W. Song, Org. Biomol. Chem. 17 (2019) 9933-9941. DOI:10.1039/C9OB02081K |
| [32] |
Y. Liao, Q. Lu, G. Chen, et al., ACS Catal. 7 (2017) 7529-7534. DOI:10.1021/acscatal.7b02558 |
| [33] |
Y. Chen, L. Liu, D. Wu, Y.P. He, A. Li, Chin. Chem. Lett. 30 (2019) 269-270. DOI:10.1016/j.cclet.2018.06.001 |
| [34] |
R. Peng, Y. Xu, Q. Cao, Chin. Chem. Lett. 29 (2018) 1465-1474. DOI:10.1016/j.cclet.2018.09.001 |
| [35] |
B.C. Boren, S. Narayan, L.K. Rasmussen, et al., J. Am. Chem. Soc. 130 (2008) 8923-8930. DOI:10.1021/ja0749993 |
| [36] |
M.M. Majireck, S.M. Weinreb, J. Org. Chem. 71 (2006) 8680-8683. DOI:10.1021/jo061688m |
| [37] |
P.C. Knutson, H.E. Fredericks, E.M. Ferreira, Org. Lett. 20 (2018) 6845-6849. DOI:10.1021/acs.orglett.8b02975 |
| [38] |
P. Wu, V.V. Fokin, Aldrichimica Acta 40 (2007) 7-17. |
| [39] |
M. Meldal, C.W. Tormøe, Chem. Rev. 108 (2008) 2952-3015. DOI:10.1021/cr0783479 |
| [40] |
J.E. Moses, A.D. Moorhouse, Chem. Soc. Rev. 36 (2007) 1249-1262. DOI:10.1039/B613014N |
| [41] |
J.E. Hein, V.V. Fokin, Chem. Soc. Rev. 39 (2010) 1302-1315. DOI:10.1039/b904091a |
| [42] |
C. Wang, D. Ikhlef, S. Kahlal, J.Y. Saillard, D. Astruc, Coordin. Chem. Rev. 316 (2016) 1-20. DOI:10.1016/j.ccr.2016.02.010 |
| [43] |
T. Henkel, R.M. Brunne, H. Müller, F. Reichel, Angew. Chem. Int. Ed. 38 (1999) 643-647. DOI:10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G |
| [44] |
A. Srivastava, L. Aggarwal, N. Jain, ACS Comb. Sci. 17 (2015) 39-48. DOI:10.1021/co500135z |
| [45] |
A. Dell'Isola, M.M.W. McLachlan, B.W. Neuman, et al., Chem. Eur. J. 20 (2014) 11685-11689. DOI:10.1002/chem.201403560 |
| [46] |
B. Banerji, S.K. Pramanik, P. Sanphui, S. Nikhar, S.C. Biswas, Chem. Biol. Drug Des. 82 (2013) 401-409. DOI:10.1111/cbdd.12164 |
| [47] |
C.S. Sevov, J.F. Hartwig, J. Am. Chem. Soc. 136 (2014) 10625-10631. DOI:10.1021/ja504414c |
| [48] |
C.S. Sevov, J.F. Hartwig, J. Am. Chem. Soc. 135 (2013) 9303-9306. DOI:10.1021/ja4052153 |