 2021, Vol. 32
2021, Vol. 32 


b School of Energy and Environment, City University of Hong Kong, Hong Kong, China;
c Department of Mechanical Engineering, The University of Melbourne, Parkville, VIC 3010, Australia;
d School of Biology and Biological Engineering, South China University of Technology, Guangzhou 510006, China;
e Institute of chemistry, Hebrew University of Jerusalem, Jerusalem 91904, Israel
Efficient explosive detection has drawn intense attentions due to increasing concerns of security [1, 2]. Especially, on-site or direct sensing of trace explosives is more appealing and essential in criminal sites and forensic analysis [3]. Compared to nitroaromatics probes that are well-developed nowadays [4, 5], detection of nitroamine that are more powerful and widely used in terrorism, e.g., RDX and HMX [6-8], are still rare due to their ultralow volatility, unfavorable reduction potentials and weak electron withdrawing abilities. In particular, HMX possesses an even lower volatility of 0.1 ppt as compared to RDX (5 ppt), making it a great challenge to probe and identify trace HMX. One of the commonly utilized method for direct HMX sensing is specialized instrument analysis, including high-performance liquid chromatography (HPLC) [9], ion mobility spectrometry [10], voltammetry technique [11, 12] and capillary electrophoresis [13]. Yet, they are not quite suitable for on-site detection. Indirect detection through decomposition-mediated strategies (e.g., radicals) [14] is another choice. The existing dilemma is, however, same reactive intermediates (NOx) are generated from decomposition of either HMX or RDX, thus hampering precise component identification and decreasing sensing selectivity. To the best of our knowledge, the direct and selective detection of trace HMX using chemical receptor has not been reported [1-14]. Thus, development of chemical probes for direct detection and identification of HMX with enhanced selectivity is still an urgent task.
Metal-organic cages (MOCs), discrete molecules assembled through coordination interactions between metal ions and organic ligands, have been recently considered as promising candidates in molecular recognition owing to their well-defined cavities and tunable functionalities [15-17]. The rational design of individual building blocks allows fine tuning of cavity size and the integration of chemical functionalities and multiple interactions inside the cavities of MOCs, facilitating the selective binding of various guests, even at single molecule level. More importantly, guest molecules binding in molecular cages could in turn induce unusual phenomena that are hardly observed in guest-free state due to the amplified host-guest events in confined environment, such as guest-induced emission amplification [18], guest-induced rearrangement [19], stabilization of reactant transition state [20], enhancement of reaction selectivity [21]. Taking advantages of these distinctive features of metal-organic cages, the rational design of ideal receptors for selective binding given target molecules should be possible.
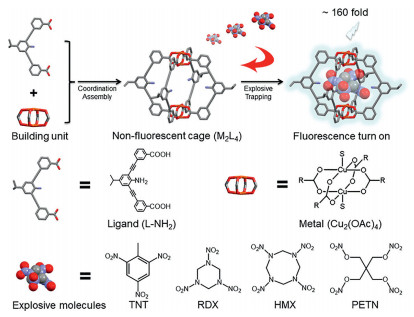
Herein we developed a non-fluorescent M2L4 molecular cage (cage A), acting as a remarkable receptor for direct and selective sensing of HMX with exceptionally high fluorescence turn-on behavior (Scheme 1). The cage A was decorated with unsaturated Cu2+ and amino groups to amplify the guest binding events and induce detectable outputs. After binding HMX, the emission of HMX@cage A complex is enhanced remarkably up to 160-folds with a detection limit of 3.5 ppb. No response to RDX and PETN, and only minute response to TNT were detected, demonstrating the unique sensing selectivity towards HMX. DFT simulation was carried out to understand the mechanism behind. Compared with the cases of TNT and RDX, DFT simulation showed that HMX exhibits the largest charge transfer to Cu2+, indicating the strongest modulation of the Cu2+single occupied molecular orbitals (SOMO), correlating well with experimental results. Therefore, the origin of such fluorescence enhancement probably is the mutual fit of both size and binding sites between host and guest, thus leading to the guest-induced perturbation of the ligand-to-metal charge-transfer (LMCT) process. To the best of our knowledge, this is the first chemical receptor for direct and selective detection of HMX based on a fluorescence turn-on approach. It should be noted that, compared to traditional turn-off MOCs explosive sensors [22-24], the turn-on approach would significantly improve the sensitivity as well as the selectivity of the probe, making it a preference for HMX detection.

|
Download:
|
| Scheme 1. Schematic illustration of the direct identification of HMX within the cage A and structures of ligand and metal (S in copper unit represents the additionally coordinated solvent molecules H2O and CH3OH), and the target molecules include nitroaromatic TNT and nitroamines RDX, HMX and PETN. | |
The ligand 3,3′-((2-amino-5-isopropyl-1,3-phenylene) bis(ethyne-2,1-diyl))dibenzoic acid (L-NH2) was synthesized from 2,6-dibromo-4-isopropylaniline (for the synthetic route, see Fig. S1 in Supporting information) to create the M2L4 cage A (Scheme 1). Through slowly layering methanol onto N,N-diethylformamide (DEF) solution that contains Cu2(OAc)4 (1 equiv.) and ligand L-NH2 (2 equiv.) at room temperature [25, 26], the molecular cage [Cu4(L-NH2)4(S)4]·xS with a yield of 80% was harvested after 5 days. Single-crystal X-ray analysis reveals the formation of a M2L4 lantern-type structure with two paddlewheel dicopper motifs bridged by four ligands L-NH2 (Fig. S2 and Table S1 in Supporting information). The distance between two Cu2+ is 9.394 Å, and the distance between two opposing aniline rings is 11.973 Å. The cage crystals are soluble and stable in DEF/dichloromethane solution. The intense peak at 1962.20 in electrospray ionization mass (ESI-MS) spectrum further confirmed the composition of [Cu4L4] + Na+ (Fig. S3 and discussion in Supporting information).
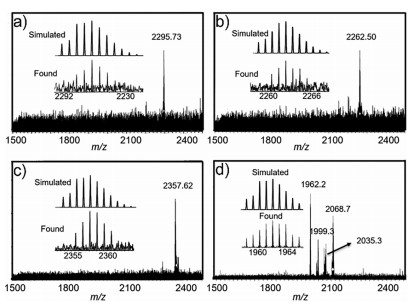
In our work, we selected two kinds of explosives as targets, including nitroaromatic TNT and nitroamines RDX, HMX and PETN (Scheme 1). The host-guest interaction between the cage A and explosive molecules was first studied by ESI-MS technique. In Figs. 1a-c, the intense peaks at m/z 2295.73, 2262.50, and 2357.62 in ESI-MS spectra were observed, which are assigned correspondingly to [Cu4L4@C7H5N3O6]·4CH3OH + H+ species, [Cu4L4@C3H6N6O6]·2H2O·2CH3OH + H+ and [Cu4L4@C4H8N8O8]·H2O·2CH3OH + K+ species, proving the 1:1 stoichiometric host-guest complexation. However, the intense peak at m/z 1962.20 in Fig. 1d shows that PETN was not encapsulated in the cage A, probably due to the steric hindrance between PETN and the cage A. However, due to the paramagnetic property of copper(Ⅱ), NMR analysis could not be performed.

|
Download:
|
| Fig. 1. ESI-MS spectra of host-guest complexes. TNT@cage A (a), RDX@cage A (b), HMX@cage A (c) and PETN@cage A (d). | |
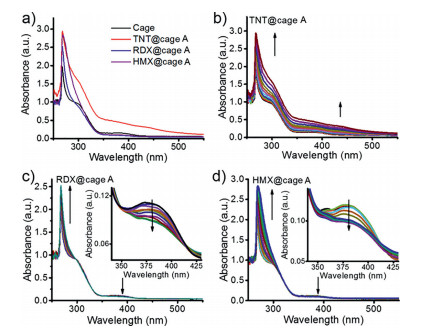
Thus, the host-guest interaction was carefully investigated and confirmed by UV–vis titration. As shown in Fig. 2a, the cage A solution exhibits obvious ligand-based charge-transfer bands at 378 nm in DEF solution. Upon addition of RDX and HMX, the intensity of absorption at visible area decreases while TNT increases the absorption at visible area, which could be attributed to the charge-transfer interaction between the amino group of cage A and TNT (Figs. 2a and b). After the addition of TNT into the solution of the cage A, the color changes from colorless to deep red, indicating the guest-binding behavior inside the cavity (Fig. S4 in Supporting information). In the case of RDX and HMX, two isosbestic points are observed at 355 nm and 413 nm, together with the decrease of the absorption peak at 378 nm (Figs. 2c and d), indicative of the prospective guest binding within the cage. The decrease of the absorption band at 378 nm might be due to the partial blockage of ligand-to-metal charge transfer (LMCT) upon interaction between HMX or RDX and metal ion center of the cage, thus leading to the strong modulation of the single occupied molecular orbitals (SOMO) of Cu2+ and further modulation of the fluorescence. The detailed mechanism would be discussed in the following context. However, the UV–vis spectra of the cage A titrated by PETN did not change due to the size-selectivity of the cage, which is consistent with the ESI-MS results. The binding affinity of TNT@cage A was calculated to be 1.43 × 104 L/mol by assuming a 1:1 binding model. For RDX and HMX, the cage A shows a moderate binding affinity of 1.03 × 104 L/mol and 1.60 × 103 L/mol, respectively (Fig. S5 in Supporting information). It should be noted that the substrate with higher binding constant does not necessarily indicate the better sensitivity. Both size- and shape-dependence determine the selectivity and sensing ability with our cage, which is an efficient sensing strategy towards selective explosive molecules trapping.

|
Download:
|
| Fig. 2. (a) The UV–vis spectra of the cage A and the formed explosive@cage A complexes in DEF solvent; (b) UV–vis spectra as obtained during the titration of the cage A (10−5 L/mol) with 10−3 L/mol TNT in DEF/CH2Cl2 solution; (c) UV–vis spectra as obtained during the titration of the cage A (10−5 L/mol) with 10−3 L/mol RDX in DEF/CH2Cl2 solution, inset shows zoomed in spectrum; (d) UV–vis spectra as obtained during the titration of the cage A (10−5 L/mol) with 10−3 L/mol HMX in DEF/CH2Cl2 solution, inset shows zoomed in spectrum. | |
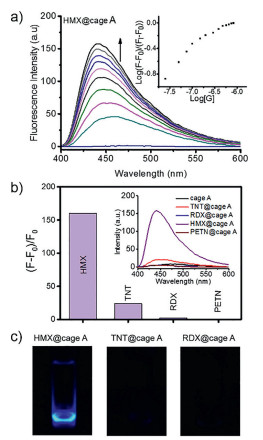
To further study the sensing ability and selectivity of the designed cage A, we investigated the fluorescence behavior of the host-guest complexes through fluorescence titration. As expected, irradiation of the cage A in pure DEF solution gave no emission due to fluorescence quenching by Cu2+ ions, while the ligand L-NH2 exhibited high fluorescence at 445 nm (Fig. S6 in Supporting information). Interestingly, high fluorescence enhancement at 445 nm could be observed upon addition of HMX into the solution of the cage, together with a blue-shift of emission (Fig. 3a). The limit of detection (LOD) was calculated to be 3.5 ppb from 3 σ/S. In contrast, the titration of the cage with TNT in DEF solution led to a little increase of the emission band at 445 nm, while RDX and PETN caused almost no change in fluorescence spectra (Fig. S7 in Supporting information). The emission band at 445 nm of HMX@cage A in DEF solution showed a significant fluorescence enhancement up to 160-folds, whereas the cage A exhibited no fluorescence response to RDX and PETN, and only minute response to TNT (Figs. 3b and c). Controlled fluorescence and absorbance experiments were also carried out that pure ligand L-NH2 was mixed with explosives (Figs. S8 and S9 in Supporting information). No fluorescence and absorbance changes were observed, indicating the crucial role of mutual fit of HMX in MOC's confined cavity to induce amplified detectable sensing signals. In Fig. 3c, the fluorescent image of HMX@cage A in DEF solution shows a significant fluorescence enhancement as compared to TNT@cage A and RDX@cage A under irradiation. The quantum yields of the cage A in the absence and presence of HMX were measured to be 0.004 and 0.062, respectively. Luminescence lifetimes were also measured. The lifetime of cage A is 4.02 ns. In the presence of HMX, the lifetime is 2.73 ns, therefore the type of luminescence is fluorescence (Figs. S10 and S11 in Supporting information). In fact, due to the designable features of metal-organic cages, cages can be facilely modified to be hydrophilic through the appropriate selection or modification of ligands or metal ions for further investigation of the sensing performance and improvement of practical applications.

|
Download:
|
| Fig. 3. (a) Emission spectra, as obtained during the titration of cage (10−7 L/mol) with 10−5 L/mol HMX in DEF solution when excited at 390 nm; inset shows the titration isotherm; (b) Fluorescence enhancement of HMX@cage A, TNT@cage A, RDX@cage A and PETN@cage A as compared to pure cage in DEF solution, inset shows the fluorescence response of the cage toward HMX, TNT, RDX and PETN after fluorescence titrations reach saturation; (c) Fluorescent images of HMX@cage A, TNT@cage A, RDX@cage A under UV light (365 nm). | |
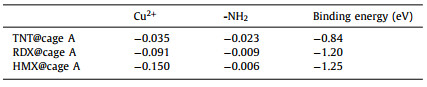
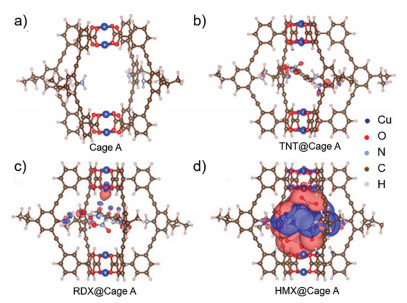
In our work, numerous attempts were made to obtain the crystal structure of host-guest complex under different conditions (e.g., solvent, temperature) to clarify host–guest interactions at molecular level, but unfortunately failed. Thus, DFT simulations were carried out to further study the inclusion of the explosives in the cage A and the possible fluorescence turn-on mechanism. The simulated geometries of the cage and host-guest complexes provide the 1:1 binary complexation (Fig. 4). In all three explosives, nitro groups are found to point toward amino groups of the cage. Particularly, in the case of HMX, two nitro groups point to amino groups and the other two nitro groups point to the Cu2+ center of the cage A (Fig. 4d). Compared with RDX and TNT, HMX induces much stronger charge transfer within the cage, and the charge transfer to Cu2+ center was much larger than that to amino groups, indicating a stronger modulation to the Cu2+ single occupied molecular orbitals (SOMO) (Table 1). In fact, the electronic structures of Cu2+ in MOC could be easily modulated through the coordination with solvent or some special molecules at the apical position in d9 Cu2+, which involved a transformation of octahedral structure [27]. After interacting with HMX, the SOMO of Cu2+ might be disturbed and the major non-radiative processes, which presumably associated with electron transfer from the organic ligand-fluorophore to the low-lying Cu2+SOMO, were turned off, thus leading to guest-induced fluorescence turn-on for HMX sensing. The blue-shift of HMX@cage A might be caused by HMX-induced conformation change of the complex structure [28]. Furthermore, X-ray photoelectron spectroscopy (XPS) measurement was performed on the sample of HMX@cage A. It was found that the valence of Cu2+ in the host-guest complexes did not change, indicating no redox reaction occurred between explosives and the cage A (Table S2 in Supporting information). This result excludes the fluorescence turn-on possibility from the Cu(Ⅰ)-based fluorophore which shows strong fluorescence. It should be noted that the calculated binding energies were performed in vacuum while the UV–vis titration was carried out in the presence of solvents. Due to the effects of solvation, the calculated and experimental binding properties cannot be compared directly.

|
Download:
|
| Fig. 4. DFT simulation of the molecular cage A and difference-electron density of different host-guest complexes. Simulated molecular M2L4 (a), difference-electron density of HMX@cage A (b), TNT@cage A (c) and RDX@cage A (d) complexes with electron accumulation represented by red and depletion by dark blue. The isosurfaces are plotted at 0.001207 e−/bohr3 for difference-election density results. | |
|
|
Table 1 Charge transfer and binding energy between the cage A and different guests. The values of charge transfer are given in electron charge. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Compared with explosive sensors reported so far, our constructed cage exhibited enhanced sensitivity and selectivity [12].
Our results indicate that through the rational design and synthesis of metal-organic cages, the cage strategy can be further developed and fulfill the requirement of direct and selective sensing various specific analytes, particularly those difficult to be detected by conventional approaches. For specific targets, the cage can be designed with multiple endo-functional sites and different cavity sizes, facilitating selective sensing. In addition, the cage can be further endowed with exo-reactive sites to couple with plasmonic nanostructures [26, 29], utilizing significantly enhanced surface plasmon to amplify the output signals and achieve higher sensitive sensing or even single-molecule recognition, which is of great importance to chemical analysis.
In summary, the first chemical receptor for direct identification of HMX with sensitivity down to ppb was developed based on the highly induced fluorescence turn-on behavior (160-fold) of a molecular cage. The unprecedented sensing features of the described receptor are attributed to synergistic effects of mutual size fit and HMX-induced perturbation of the ligand-to-metal charge-transfer (LMCT) process. With the very promising results achieved, we believe our work would establish a new approach for direct and selective detection of trace HMX, and the findings in this work could promote new sensing mechanism for developing fluorescent materials with remarkable sensing properties.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was made possible as a result of a generous grant from the National Natural Science Foundation of China (NSFC, Nos. 21773135, 22032003, 21821001), the Ministry of Science and Technology (MOST, No. 2017YFA0204501), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, No. TRR61). The authors wish to acknowledge Professor Wei Zhu for his helpful discussions regarding the experimental design.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.05.051.
| [1] |
W.P. Lustig, S. Mukherjee, N.D. Rudd, et al., Chem. Soc. Rev. 46 (2017) 3242-3285. DOI:10.1039/C6CS00930A |
| [2] |
X. Sun, Y. Wang, Y. Lei, Chem. Soc. Rev. 44 (2015) 8019-8061. DOI:10.1039/C5CS00496A |
| [3] |
H. Wang, W.P. Lustig, J. Li, Chem. Soc. Rev. 47 (2018) 4729-4756. DOI:10.1039/C7CS00885F |
| [4] |
S.W. Thomas, G.D. Joly, T.M. Swager, Chem. Rev. 107 (2007) 1339-1386. DOI:10.1021/cr0501339 |
| [5] |
M.E. Germain, M.J. Knapp, Chem. Soc. Rev. 38 (2009) 2543-2555. DOI:10.1039/b809631g |
| [6] |
M. Riskin, R. Tel-Vered, I. Willner, Adv. Mater. 22 (2010) 1387-1391. DOI:10.1002/adma.200903007 |
| [7] |
Z. Hu, K. Tan, W.P. Lustig, et al., Chem. Sci. 5 (2014) 4873-4877. DOI:10.1039/C4SC02157F |
| [8] |
L. Mosca, S. Karimi Behzad, P. Anzenbacher, et al., J. Am. Chem. Soc. 137 (2015) 7967-7969. DOI:10.1021/jacs.5b04643 |
| [9] |
H.D. Craig, T.F. Jenkins, M.T. Johnson, et al., Talanta 198 (2019) 284-294. DOI:10.1016/j.talanta.2019.02.005 |
| [10] |
M. Najarro, M.E. Dávila Morris, M.E. Staymates, R. Fletcher, G. Gillen, Analyst 137 (2012) 2614-2622. DOI:10.1039/c2an16145a |
| [11] |
Y. Xu, W. Lei, Z. Han, et al., Electrochim. Acta 216 (2016) 219-227. DOI:10.1016/j.electacta.2016.09.010 |
| [12] |
Ş. Saǧlam, A. Üzer, E. Erçaǧ, R. Apak, et al., Anal. Chem. 90 (2018) 7364-7370. DOI:10.1021/acs.analchem.8b00715 |
| [13] |
C.A. Groom, A. Halasz, L. Paquet, P. D'Cruz, J. Hawari, J. Chromatogr. A 999 (2003) 17-22. DOI:10.1016/S0021-9673(03)00389-3 |
| [14] |
C. Wang, H. Huang, B.R. Bunes, et al., Sci. Rep. 6 (2016) 25015. DOI:10.1038/srep25015 |
| [15] |
A.J. McConnell, C.S. Wood, P.P. Neelakandan, J.R. Nitschke, Chem. Rev 115 (2015) 7729-7793. DOI:10.1021/cr500632f |
| [16] |
N. Hosono, S. Kitagawa, Acc. Chem. Res. 51 (2018) 2437-2446. DOI:10.1021/acs.accounts.8b00361 |
| [17] |
M.D. Ward, C.A. Hunter, N.H. Williams, Acc. Chem. Res. 51 (2018) 2073-2082. DOI:10.1021/acs.accounts.8b00261 |
| [18] |
K. Hagiwara, M. Otsuki, M. Akita, M. Yoshizawa, Chem. Commun. 51 (2015) 10451-10454. DOI:10.1039/C5CC02931G |
| [19] |
D. Preston, A. Fox-Charles, W.K.C. Lo, J.D. Crowley, Chem. Commun. 51 (2015) 9042-9045. DOI:10.1039/C5CC02226F |
| [20] |
M. Yoshizawa, M. Tamura, M. Fujita, Science 312 (2006) 251-255. DOI:10.1126/science.1124985 |
| [21] |
J. Wei, L. Zhao, C. He, et al., J. Am. Chem. Soc. 141 (2019) 12707-12716. DOI:10.1021/jacs.9b05351 |
| [22] |
Y. Han, H.V. Huynh, Dalt. Trans. 40 (2011) 2141-2147. DOI:10.1039/C0DT01037E |
| [23] |
S. Shanmugaraju, H. Jadhav, Y.P. Patil, P.S. Mukherjee, Inorg. Chem. 51 (2012) 13072-13074. DOI:10.1021/ic301716y |
| [24] |
K. Acharyya, P.S. Mukherjee, Chem. Commun. 50 (2014) 15788-15791. DOI:10.1039/C4CC06225F |
| [25] |
J.R. Li, H.C. Zhou, Nat. Chem. 2 (2010) 893-898. DOI:10.1038/nchem.803 |
| [26] |
C. Wang, J. Shang, Y. Lan, et al., Adv. Funct. Mater. 25 (2015) 6009-6017. DOI:10.1002/adfm.201503071 |
| [27] |
Z. Li, N. Kishi, K. Yoza, M. Akita, M. Yoshizawa, Chem. Eur. J. 18 (2012) 8358-8365. DOI:10.1002/chem.201200155 |
| [28] |
M. Zhang, G. Feng, Z. Song, et al., J. Am. Chem. Soc. 136 (2014) 7241-7244. DOI:10.1021/ja502643p |
| [29] |
C. Wang, L. Tian, W. Zhu, et al., Chem. Sci. 9 (2018) 889-895. DOI:10.1039/C7SC03536E |