 2021, Vol. 32
2021, Vol. 32 


Aminoboranes containing a B-N σ bond are an important class of compounds that are used to construct novel BN-based polymeric materials [1]. In organic synthetic chemistry, they also serve as efficient aminoboration reagents for a variety of unsaturated molecules, e.g., isocyanates [2], carbodiimides [3], aldehydes [4], enones [5] and alkynes [6]. Primary aminoboranes have also been explored as ligand precursors to stabilize highly-reactive low-coordinated metal complexes by taking advantage of the π-interactions between the lone pair of electrons on nitrogen and the empty p orbital of boron [7]. Thus, the development of an efficient method to synthesize aminoboranes is of great significance.
In early reports, aminoboranes were obtained through cleavage of Si-N or Sn-N bonds by boranes or haloboranes [8]. Conventional salt elimination reactions of lithium amides with BCl3 also provided the target compounds [9]. The selective hydroboration of imines provides an atom-economic pathway to obtain aminoboranes [10]. Recently, dehydrogenative coupling of amines and boranes to aminoboranes has emerged and drawn increasing attention because it employs readily-available starting materials and releases H2 as the only side product. Although it was reported that the dehydrogenation of amines and boranes occasionally occurs under catalyst-free conditions [11], the reactions can be significantly promoted using a suitable catalytic system. Hill and co-workers disclosed that a β-diketiminato magnesium butyl complex 1 (Fig. 1) enabled the dehydrocoupling of pinacolborane (HBpin) or 9-borabicyclo[3.3.1]nonane (9-BBN) with a range of amines [12]. Roesky et al. prepared an aluminum dihydride 2 that catalyzed the dehydrocoupling of boranes and amines [13]. Alkali metal (Li, Na and K) hexamethyldisilazides 3 were also found to be active catalysts for such dehydrogenation reactions by Panda's group [14]. Power et al. used group 14 tin(Ⅱ) methoxides 4 for catalytic dehydrocoupling reactions [15]. However, the substrate scope of boranes in all of the above dehydrogenation reactions was limited to cyclic secondary boranes, i.e., HBcat (catechol borane), HBpin, and 9-BBN, and most of the above catalysts exhibited only low to moderate activities.

|
Download:
|
| Fig. 1. Reported catalysts for the dehydrocoupling reactions of boranes and amines. | |
{kind=link}
In our previous work, we demonstrated that the scandocene alkyl complex Cp*2ScCH2SiMe3 (5, Cp* = C5Me5) [16] was a highly efficient catalyst for the dehydrogenation of dimethylamine-borane to cyclic borazine [17]. Mechanistic studies indicated that rapid β-H elimination to the corresponding scandium hydride played a crucial role in the catalytic reaction. Encouraged by this result, herein, we further investigated scandocene alkyl complex 5 for the intermolecular dehydrocoupling of boranes and amines. Gratifyingly, non-cyclic and primary boranes were successfully applied for the first time, affording a new family of aminoboranes under mild conditions. A plausible mechanism was also proposed based on the isolation and characterization of catalytic intermediates from the corresponding stoichiometric reactions.
We initially examined the reaction of a non-cyclic secondary borane, dicyclohexylborane (6, HBCy2), with aliphatic n-propylamine 7a in C6D6 solution. The reaction rapidly generated a new resonance at δ −0.8 ppm in the 11B NMR spectrum, indicating the formation of a Lewis acid-base adduct Cy2BH·H2NnPr that did not undergo self-dehydrogenation for up to 24 h at room temperature. Nevertheless, H2 elimination readily occurred and was complete within 15 min at room temperature upon the addition of a catalytic amount of Sc complex 5 (5 mol%). The resulting aminoborane 8a features a broad 11B NMR signal at δ 45.6 ppm, which is significantly shifted to lower-field, indicating the presence of tricoordinate boron. In 1H NMR spectrum, a signal for NH was observed at δ 3.67 ppm. The addition of an excess amount of HBCy2 did not lead to further dehydrogenation even under relatively harsh conditions (10 h, 60 ℃). Subsequently, reactions of HBCy2 with a variety of amine substrates were investigated under such conditions (5 mol% 5, 6/7 = 1, in C6D6, room temperature; Scheme 1). Detailed isolations and characterizations of corresponding products were provided in Supporting information.

|
Download:
|
| Scheme 1. Dehydrocoupling of HBCy2 and amines catalyzed by Sc complex 5. Conditions: [6] = 0.1 mmol, [7] = 0.1 mmol, 5 mol% 5. Reactions were performed in C6D6 at room temperature in an unsealed vial. Yields were determined by 1H NMR using ferrocene as an internal standard. aReaction was conducted at 80 ℃. b0.2 mmol 6 was used. | |
{kind=link}
Several aliphatic primary amines were applicable for the dehydrogenation and completely transformed into the expected aminoboranes 8b-8e within a short time (15–40 min). Amines containing oxygen atoms were compatible with the catalyst system. Cyclohexylamine required a long time (4 h) to achieve full conversion to the corresponding aminoborane 8f. Dehydrocoupling reactions were greatly facilitated when using aromatic amines as the substrates, e.g., the production of 8g-8i. Notably, reaction of para-bromoaniline with HBCy2 gave 8h in quantitative yield in only 2 min. The molecular structure of compound 8i was further confirmed by single-crystal X-ray diffraction and showed a planar tricoordinate geometry at boron (for details, see Supporting information). 2, 6-Dimethylaniline was completely converted into the corresponding dehydrogenated product 8j in a prolonged reaction time (12 h) under heating (80 ℃), presumably due to severe steric hindrance. When using para-acetylaniline as the substrate, hydroboration of the carbonyl group was preferred. Nevertheless, the dehydrocoupling product 8k was obtained by adding 2 equiv. borane 6. Secondary amines, e.g., morpholine and tetrahydropyrrole, were also suitable for this reaction, affording 8l and 8m, respectively, albeit under relatively harsh conditions (2 h, 80 ℃). No dehydrogenation product was observed when treatment of sterically hindered (Me3Si)2NH with HBCy2 under the same conditions.
Reactions of primary thexylborane 9 with a wide range of amines were also investigated under the optimized conditions (5 mol% 5, 9/7 = 1, in C6D6, room temperature; Scheme 2). Again, a control reaction of thexylborane with n-propylamine 7a afforded a Lewis acid-base adduct, which did not decompose within 12 h at 60 ℃. In the presence of scandium catalyst 5, the resultant adduct rapidly underwent dehydrogenation to form aminoborane 10a. Further dehydrogenation of 10a to form a B=N double bond was not observed, even when reacted overnight at 60 ℃. Compound 10a showed a typical tricoordinate borane resonance at δ 44.8 ppm in the 11B NMR spectrum as a doublet with a 1JHB coupling constant of 126.5 Hz. Increasing the amount of either borane or amine did not alter the reaction outcome. A series of aliphatic and aromatic primary amines was involved and gave the corresponding aminoboranes 10b-10k in a short time. It is noteworthy that cyclic secondary amines generated dehydrogenation products 10l and 10m under mild conditions (5–10 min, room temperature), in sharp contrast to the reactions of HBCy2 with secondary amines. A double dehydrogenation product 10n could be obtained when treatment of 1,4-diaminobutane with 2 equiv. thexylborane.

|
Download:
|
| Scheme 2. Dehydrocoupling of thexylborane and amines catalyzed by Sc complex 5. Conditions: [9] = 0.1 mmol, [7] = 0.1 mmol, 5 mol% 5. Reactions were performed in C6D6 at room temperature in an unsealed vial. Yields were determined by 1H NMR using ferrocene as an internal standard. aReaction was conducted in the presence of 2.5 mol% 5. bReaction was conducted at 60 ℃. | |
{kind=link}
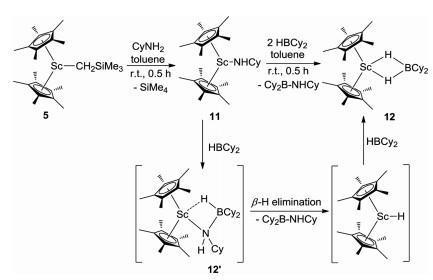
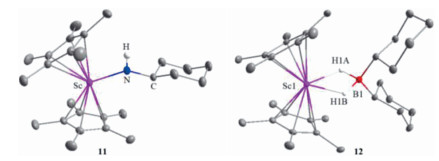
To gain further insight into the Sc-catalyzed dehydrocoupling reactions, several stoichiometric reactions were conducted. The reaction of Sc alkyl complex 5 with an equimolar amount of amine CyNH2 gave the expected Sc amide complex 11 in 92% yield (Scheme 3) with the release of SiMe4. Complex 11 was comprehensively characterized by NMR spectroscopy, elemental analysis, and single-crystal X-ray diffraction (Fig. 2). The bond length of Sc1-N1 was found to be 2.0112(16) Å, which was comparable to those of previously-reported complexes containing Sc-NHR moieties [18]. Further reaction of complex 11 with 1 equiv. of HBCy2 rapidly yielded the dehydrocoupling product 8f and a new species with a 11B NMR signal at δ 8.0 ppm. In addition, the unreacted complex 11 was also detected in a 1:1 ratio with 8f in the reaction mixture, suggesting that 2 equiv. of HBCy2 were required for this reaction. The new species was finally isolated in 65% yield and identified as the HBCy2 coordinated scandium hydride 12 (Scheme 3) by single-crystal X-ray diffraction. The molecular structure of complex 12 was shown in Fig. 2. Hydride atoms were located in a Fourier difference map and refined with isotropic displacement parameters. Two hydride ligands were bound to the metal center with bond lengths of 1.97(1) Å and 2.02(1) Å, respectively. The Sc…B distance was found to be 2.634(2) Å, which is significantly longer than those in previously-reported scandium tetrahydridoborate complexes [19]. It is noted that the three-component reaction of scandium alkyl complex 5, CyNH2, and HBCy2 also led to identical results. Therefore, it is likely that the scandium amidoborane intermediate 12′ was formed initially as in our previous case [17], which underwent rapid β-H elimination to afford scandocene hydride and aminoborane. The resultant metal hydride was highly reactive and thus immediately trapped by hydroborane. Unsuccessful isolation of complex 12′ presumably resulted from the sterically bulky amino group.

|
Download:
|
| Scheme 3. Preparation of complexes 11 and 12. | |
{kind=link}

|
Download:
|
| Fig. 2. Molecular structures of complexes 11 and 12. Hydrogen atoms (except NH and BH) are omitted for clarity, and ellipsoids are drawn at the 30% probability level. Selected bond lengths (Å) and angles (deg): 11: Sc-N, 2.0112(16); N-C, 1.453(2); Sc-N-C, 147.42(14); 12: Sc1-H1A, 2.02(1); Sc1-H1B, 1.97(1); B1-H1A, 1.24(1); B1-H1B, 1.28(9); H1A-Sc1-H1B, 53.8(8); H1A-B1-H1B, 91.6(13). | |
{kind=link}
Both isolated complexes 11 and 12 were employed as catalysts for the reaction of HBCy2 with CyNH2 under our typical conditions. Scandium amide complex 11 showed comparable activity to that of parent complex 5. In contrast, the catalytic activity drastically decreased when using complex 12 as a catalyst, indicating that 12 should not be included in the catalytic cycle of Sc-catalyzed dehydrocoupling reaction. In addition, the reaction of HBCy2 with CyNH2 in a 10:1 molar ratio catalyzed by scandocene alkyl complex 5 did not yield any dehydrocoupling product under the same conditions, suggesting that excess borane inhibited the reaction. These experimental results were consistent with those observed in main-group metal Mg-catalyzed dehydrocoupling of boranes with amines [12].
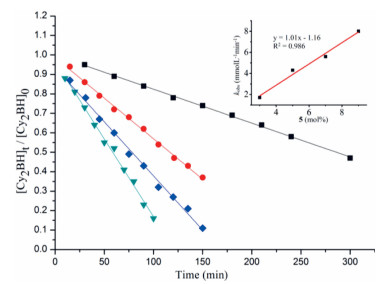
Kinetic studies of the dehydrocoupling reaction of HBCy2 with CyNH2 were also conducted by varying the catalyst loading from 3 mol% to 9 mol% and monitored by NMR spectra (for details, see Supporting information). As shown in Fig. 3, plots of the substrate conversion versus reaction time for all trials generated straight lines, indicating a zero-order kinetic behavior. Based on these results, a first-order dependence on catalyst 5 was observed (Fig. 3). Replacement of HBCy2 with deuterated DBCy2 resulted in a significant kinetic isotope effect (KIE) of 4.0 (Fig. S88 in Supporting information). In contrast, deuterated amine only afforded a KIE of 1.2. These results suggested that B-H bond cleavage might be involved in the rate-determining step.

|
Download:
|
| Fig. 3. Plot of [Cy2BH]t /[Cy2BH]0 versus time at various catalyst loadings. Standard substrate concentration (0.67 mol/L): 3 mol% 5 (black squares, y = −0.0017x + 0.998, R2 = 0.998); 5 mol% 5 (red dots, y = −0.0043x + 0.992, R2 = 0.997); 7 mol% 5 (blue diamonds, y = −0.0056x + 0.934, R2 = 0.994); 9 mol% 5 (green triangles, y = −0.0080x + 0.967, R2 = 0.995). Inset: first-order plot for [5]. | |
{kind=link}
Based on the above observations and previous literature reports, a plausible mechanistic framework for the Sc-catalyzed dehydrocoupling of boranes and amines is shown in Supporting information (Fig. S89 in Supporting information). The reaction of Sc alkyl complex 5 with an amine gave a Sc amide complex with the release of SiMe4, which easily reacted with borane through N-B and Sc-H interactions to form a Sc amidoborane intermediate. Rapid β-H elimination then occurred to produce the aminoborane product and Sc hydride. Finally, the dehydrogenation of Cp*2ScH with an amine produced Sc amide to complete the catalytic cycle. Alternatively, Sc hydride species could be also trapped by hydroborane.
In summary, the catalytic dehydrocoupling of non-cyclic boranes, i.e., dicyclohexylborane and thexylborane, with a wide range of amines has been achieved for the first time by using a scandocene alkyl complex under mild conditions. The reactions provided a new family of aminoboranes with a high atom economy and broad substrate scope. Mechanistic studies, including stoichiometric and kinetics experiments, indicated that a scandium hydride derived from the β-H elimination of the corresponding metal amidoborane played a critical role in catalysis. Exploration of the applications of such aminoboranes in synthetic chemistry is in progress.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (No. 21871204).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.05.041.
| [1] |
(a) A. Staubitz, A.P.M. Robertson, Sloan M.E, I. Manners, Chem. Rev. 110 (2010) 4023-4078; (b) A.L. Colebatch, A.S. Weller, Chem. Eur. J. 25 (2019) 1379-1390; (c) X. Long, C. Dou, J. Liu, L. Wang, Chin. Chem. Lett. 29 (2018) 1343-1346; (d) L. Li, Y. Gao, C. Dou, J. Liu, Chin. Chem. Lett. 31 (2020) 1193-1196. |
| [2] |
R.H. Cragg, M.F. Lappert, B.P. Tilley, J. Chem. Soc. (1964) 2108-2115. DOI:10.1039/jr9640002108 |
| [3] |
R. Jefferson, M.F. Lappert, B. Prokai, B.P. Tilley, J. Chem. Soc. A (1966) 1584-1590. |
| [4] |
(a) M. Suginome, L. Uehlin, M. Murakami, J. Am. Chem. Soc. 126 (2004) 13196-13197; (b) G.P. Junor, E.A. Romero, X. Chen, R. Jazzar, G. Bertrand, Angew. Chem. Int. Ed. 58 (2019) 2875-2878. |
| [5] |
C. Solé, E. Fernández, Angew. Chem. Int. Ed. 125 (2013) 11561-11565. DOI:10.1002/ange.201305098 |
| [6] |
(a) E. Chong, S.A. Blum, J. Am. Chem. Soc. 137 (2015) 10144-10147; (b) K. Yuan, S. Wang, Org. Lett. 19 (2017) 1462-1465. |
| [7] |
(a) R.A. Bartlett, X. Feng, M.M. Olmstead, P.P. Power, K.J. Weese, J. Am. Chem. Soc. 109 (1987) 4851-4854; (b) G. Linti, J. Organomet. Chem. 465 (1994) 79-83; (c) H. Chen, R.A. Bartlett, M.M. Olmstead, P.P. Power, S.C. Shoner, J. Am. Chem. Soc. 112 (1990) 1048-1055; (d) M. Aman, O. Mrózek, L. Dostál, Z. Růžičková, R. Jambor, J. Organomet. Chem. 872 (2018) 1-7. |
| [8] |
(a) J.F. Janik, C.K. Narula, E.G. Gulliver, E.N. Duesler, R.T. Paine, Inorg. Chem. 27 (1988) 1222-1227; (b) B. Wrackmeyer, B. Schwarze, W. Milius, J. Organomet. Chem. 489 (1995) 201-205; (c) B. Wrackmeyer, G. Kehr, S.Z. Ali, Z. Naturforsch. 53b (1998) 393-395; (d) B. Wrackmeyer, H.E. Maisel, M. Herberhold, J. Organomet. Chem. 637 (2001) 727-732. |
| [9] |
(a) Y. Tang, X. Li, PT CN 101440101, 2009; (b) Z. Gao, X. Li, PT CN 102093399, 2011. |
| [10] |
C.C. Chong, R. Kinjo, ACS Catal 5 (2015) 3238-3259. DOI:10.1021/acscatal.5b00428 |
| [11] |
(a) E.A. Romero, J.L. Peltier, R. Jazzar, G. Bertrand, Chem. Commun. 52 (2016) 10563-10565; (b) C.M. Vogels, P.E. O'Connor, T.E. Phillips, et al., Can. J. Chem. 79 (2001) 1898-1905. |
| [12] |
D.J. Liptrot, M.S. Hill, M.F. Mahon, A.S.S. Wilson, Angew. Chem. Int. Ed. 54 (2015) 13362-13365. DOI:10.1002/anie.201505949 |
| [13] |
Z. Yang, M. Zhong, X. Ma, et al., J. Am. Chem. Soc. 138 (2016) 2548-2551. DOI:10.1021/jacs.6b00032 |
| [14] |
A. Harinath, S. Anga, T.K. Panda, RSC Adv. 6 (2016) 35648-35653. DOI:10.1039/C6RA04125F |
| [15] |
J.D. Erickson, T.Y. Lai, D.J. Liptrot, M.M. Olmstead, P.P. Power, Chem. Commun. 52 (2016) 13656-13659. DOI:10.1039/C6CC06963K |
| [16] |
M.A. St. Clair, B.D. Santarsiero, J.E. Bercaw, Organometallics 8 (1989) 17-22. DOI:10.1021/om00103a004 |
| [17] |
P. Xu, X. Xu, Organometallics 38 (2019) 3212-3217. DOI:10.1021/acs.organomet.9b00461 |
| [18] |
(a) X. Han, L. Xiang, Y. Chen, Chem. Eur. J. 23 (2017) 14728-14732; (b) J. Chu, X. Han, Y. Chen, et al., J. Am. Chem. Soc. 136 (2014) 10894-10897; (c) J. Chu, Q. Zhou, Y. Li, X. Leng, Y. Chen, Sci. China Chem. 57 (2014) 1098-1105. |
| [19] |
(a) F. Bonnet, C.D.C. Violante, M. Visseaux, Chem. Commun. (2009) 3380-3382; (b) B.G. Segal, S.J. Lippard, Inorg. Chem. 17 (1978) 844-850. |