 2021, Vol. 32
2021, Vol. 32 


Transition metal-catalyzed heteroatom-direct C-H activation and functionalization has emerged as one of the most efficient and powful methods for creating new carbon-carbon bonds in organic synthesis [1-4]. Nitrogen functional groups (e.g., amines, imines, amides and nitriles) and oxygen functional groups (e.g., ketones and carboxylates) are the mainly established directing groups due to their suitable coordination ability to metal center. In this context, palladium-catalyzed δ-site C−H activation of aliphatic amines has been less well developed compared with the corresponding γ-site, which may be a kinetically less favorable six-membered palladacycle intermediate [5, 6]. Recently, the δ-C(sp3)−H functionalization of alkyl picolinamides, picolinamide-protected amino acids and peptides, and picolinamides has been realized by Chen group, Daugulis group, Shi group and Wang group [7-10].
Free amines are challenge in palladium-catalyzed direct C−H activation reactions as a result of the formation of the stable bis-amine palladium complexes [11]. More recently, Bannister and coworkers reported the Pd(II)-catalyzed δ-C(sp3)−H arylation of free primary aliphatic amines using the unmodified NH2 group as directing group (Scheme 1) [12]. This notable reaction presents a significant advance in the Pd-catalyzed C-H activation chemistry. Herein, the catalytic activity of the reaction has attracted our attention. It is intriguing but puzzling that the complete loss of reactivity occurs in the absence of Ag(I) additives (Scheme 1). This indicates that the silver(I) additives are vital in the catalytic cycle than merely iodide abstraction, or the reaction would give a catalytic amount of products. Although the formation of a palladium-silver heterodimer in the transition state might elucidate this phenomenon [13-15], the mechanism of this important reaction has not been explored. In view of that the silver(I) salt is the most commonly used additive in palladium catalysis, it is of importance to disclose whether it plays a more active role in the full catalytic cycle than just abstracting iodine in this reaction system.

|
Download:
|
| Scheme 1. Palladium-catalyzed C(sp3)−H arylation of amines. | |
{kind=link}
To unveil the origins of catalytic activity and identify the crucial role the silver(I) additives in this important catalytic reactions, we carry out this theoretical and experimental study. In the light of density functional theory (DFT) calculations and mass spectrometry analyses via control experiments, a kinetically favored Pd-Ag heterodimeric C-H activation and C-C coupling mechanism was revealed and the active heterodimeric Pd-Ag species were detected. The mechanistic findings demonstrate the multifunctional role of Ag(I) additives on supporting the activation and formation of all chemical bonds in the catalytic cycle.
The reaction in Scheme 1 was selected as a representative to explore the mechanism and origin of silver(I)-effect activity in the palladium-catalyzed C(sp3)-H arylation of amine 1a. The most stable form of Pd(II) acetate is trimer, which should be initiated to produce the active species. The Pd(II) acetate can undergo dissociation and substrate coordination to form the bis-amine Pd(II) intermediate IN1. The initiation conversion is thermodynamically favorable by 21.8 kcal/mol (1/3 Pd3(OAc)6 + 2 1a → IN1; ΔG0 = -21.8 kcal/mol) as a result of the strong coordination ability of N atom to Pd(II) center. This result agrees with the experimental observation that the bis-Pd(II) intermediate IN1 was obtained using amine 1a and Pd(OAc)2 in chloroform [12]. Hence, IN1 is deemed as the catalyst resting state to construct the free energy profiles.
The mechanism of palladium(II)/acetate-catalyzed C-H activation have been recognized as a concerted metallation-deprotonation (CMD) process [16-20]. This has guided us to locate the reaction pathway for the C‒H activation of amine 1a starting at IN1 (Fig. 1). IN1 dissociates one molecular of substrate 1a and gives the intermediate IN2, wherein one of the acetate ligand binds to the Pd(II) center in an κ2-mode. The dissociation step requires a barrier of 27.3 kcal/mol (TS1) and is energetically unfavorable by 25.9 kcal/mol owing to the loss of NH2-containing coordination reactant. IN2 then proceeds via the six-membered CMD transition state TS2, in which the acetate acts as an internal base to activate the C(sp3)−H bond, affording the cyclopalladate IN3. The optimized structures of TS2 are shown in Fig. 1. TS2 is 35.7 kcal/mol relative to IN1, the energy barrier of which is too high to explain the catalytic activity (Scheme 1). The computational results discussed above stimulate us to explore other C(sp3)−H activation pathways assisted by the silver(I) additives with lower barriers.

|
Download:
|
| Fig. 1. Free energy profiles of C-H activation via monomeric and heterodimeric mechanism, and the optimized geometries for TS2 and TS3. Selected bond lengths are labeled in Å. | |
{kind=link}
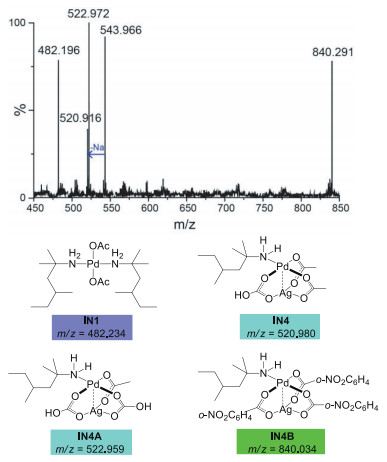
Experimentally, the Ag(I) additive is indispensable for the C(sp3)−H arylation. When the Ag(I) additive is removed from the system, the reaction is completely inactivated (Scheme 1). This implies that Ag(I) may play other roles besides iodide abstraction in the catalytic cycle. Herein, we considered the insertion of Ag(I) salt into the Pd(II) center and located the heterodimeric Pd-Ag intermediate IN4 (Fig. 1) [21]. In IN4, the distance of Pd-Ag bond is 2.75 Å, which is analogous to the observed Pd(II)-Ag(I) bond experimentally. This indicates a clearly heterometallic d8-d10 bonding interaction [22, 23]. The electronic influence together with the bridging effects of acetate ligands explains the stability of IN4. IN4 then proceeds to the C‒H activation and generates IN5 via TS3, wherein the bicarbonate connected to Ag(I) center serves as the base to break the C‒H bond. TS3 necessitates an energy barrier of 20.1 kcal/mol relative to IN1, which is feasible under the experimental condition. Herein, the heterometallic Pd(II)-Ag(I) bonding interaction increases the electric density of Pd(II) center and reduces its eletrophilicity, which is responsible for the higher elementary reaction barrier of TS3 (17.4 kcal/mol) compared with TS2 (9.8 kcal/mol). Note that, the barrier of TS2 via monomeric mechanism is disfavored due to the energy consumption of the transformation from IN1 to IN2 that makes its total energy barrier up to 35.7 kcal/mol. Although the elementary reaction barrier of TS3 (17.4 kcal/mol) is higher than that of TS2 (9.8 kcal/mol), the stability of active species IN4 makes the heterometallic C-H activation kinetically feasible.
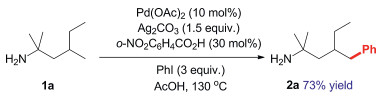
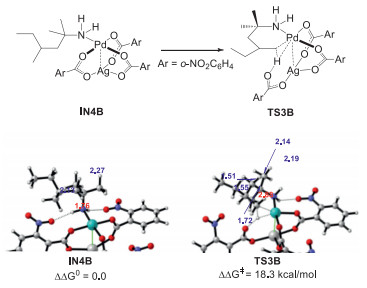
Based on the computational results, the heterodimeric Pd-Ag intermediate IN4 is only endergonic by 2.7 kcal/mol relative to the catalyst resting state IN1. Considering the small energy cost, we carried out mass spectrometry (MS) experiments to detect the possible active catalyst IN4. A 2:1:1:0.5 mixture of amine 1a, Pd(OAc)2, Ag2CO3, o-nitrobenzoic acid was dissolved in AcOH solvent and analyzed by high-resolution mass spectrometry (MALDI-TOF MS). Three peaks assigned to dimeric Pd−Ag species IN4, IN4A and IN4B were observed (Fig. 2), wherein IN4A and IN4B could be the replaced products of IN4 with HCO3- and o-NO2-PhCO2- in solvent. These heterodimeric Pd-Ag complexes could all serve as the real active species in the catalytic system. Note that, the catalyst resting state IN1 was also observed in the MS analysis, which is consistent with the experimental results by Bannister et al. [12]. The experimental evidences together with the computational results provide relatively reliable proofs for the heterodimeric Pd−Ag species in this Pd-catalyzed C‒H activation reaction with Ag(I) additives. Fig. 3 presents the energies barrier of the C‒H activation step via the heterodimeric Pd−Ag species IN4B, wherein the calculated result indicates that this pathway is kinetically attainable. Herein, the electron-withdrawing ligand can increase the eletrophilicity of the Pd(II) center, which avails the formation of Pd−C bond to promote the C − H activation in this catalytic system. In addition, the hydrogen bonding interactions between the NH2 moiety and o-NO2-PhCO2 ligand can stabilize the heterodimeric species IN4B and further promote this pathway. This may explain that the higher yield (73%) was obtained with the addition of 2-nitrobenzoic acid experimentally [12]. Overall, the computational studies and mass spectrometry analysis carried out above illuminate that the Pd(OAc)2 is the primary catalyst, the bis-amine Pd(II) complex IN1 acts as the catalyst resting state, but the heterodimeric Pd−Ag species IN4 and its analogues work as the actually active catalyst.

|
Download:
|
| Fig. 2. MALDI-TOF-MS showing peaks assigned to [IN1]+, [IN4]+, [IN4A]+, [IN4 + Na]+, and [IN4B]+. | |
{kind=link}

|
Download:
|
| Fig. 3. Optimized geometries of IN3 and TS3B. | |
{kind=link}
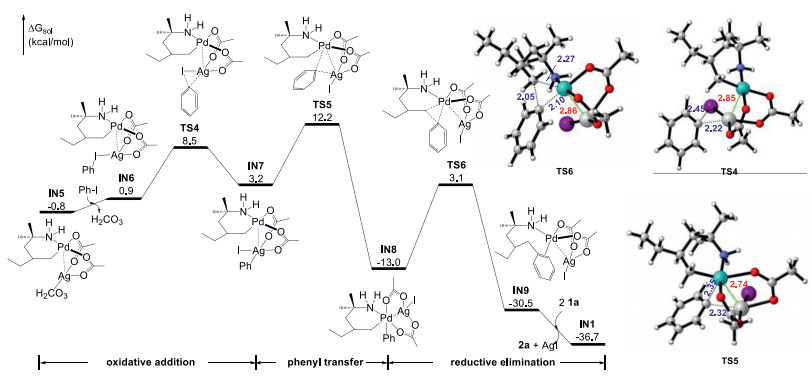
After C-H activation, the reaction will undertake oxidative addition to cleave pH-I bond and reductive elimination to build new C-C bond [24-26]. Herein, we considered the C-C coupling via heterodimeric pathway (Fig. 4), which was demonstrated to be kinetically favored than that via monomeric Pd(II) pathway (see Supporting information for details). Initially, IN5 suffers substitution to form the PhI-coordinated complex IN6, wherein pH-I adds to the Ag(I) center via oxidative transition state TS4 (ΔG≠ = 9.3 kcal/mol) offering intermediate IN7. Complex IN7 then proceeds via phenyl transfer from Ag center to Pd center via TS5 (ΔG≠ = 13.0 kcal/mol) to facilitate the subsequent C-C bond formation, which involves a formal two-electron transfer leading to Pd-Ag complex IN8. C-C reductive elimination occuring via TS5 (ΔG≠ = 16.1 kcal/mol) generates the Pd-Ag intermediate IN9 and finishes the arylation. This step is thermodynamically favorable by 17.5 kcal/mol. What is more, we calculated the oxidative addition of pH-I to the Pd(II) center via monomeric Pd pathway (TS7, ΔG≠ = 32.2 kcal/mol), which is 23.7 kcal/mol higher than TS4 (see Supporting information for details). Finally, IN9 reacts with two 1a by ligand exchange to regenerate IN1 and release 2a and byproduct AgI.

|
Download:
|
| Fig. 4. Free energy profile of the palladium-catalyzed C-C coupling aided by silver additive, and the optimized geometries of TS4, TS5 and TS6. Selected bond lengths are labeled in Å. | |
{kind=link}
In summary, we have explored the detailed mechanism of the C(sp3)-H arylation of amines enabled by palladium catalysis with silver(I) additives by virtue of a combined computational and experimental study. Based on the mechanistic investigation, the catalytic cycle consists of C-H activation, oxidative addition, phenyl transfer and reductive elimination to provide the arylated product. In contrast to the common monomeric Pd mechanism, all these steps take place via the heterodimeric Pd-Ag manner. Experimentally, the active heterodimeric Pd-Ag species were detected by mass spectrometry, which testifies the heterodimeric pathway. Overall, the theoretical and experimental studies provide new insights into the mechanism of Pd(II)-catalyzed C(sp3)‒H arylation reactions with Ag(I) additives, which reveals the synergistic manner of palladium catalysis and Ag(I) additives. The mechanistic insights endow useful implications for the further development of catalytic C-H functionalization chemistry.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Tianjin University and the National Natural Science Foundation of China (Nos. 22073067 and 21673156).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.04.044.
| [1] |
X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Angew. Chem. Int. Ed. 48 (2009) 5094-5115. DOI:10.1002/anie.200806273 |
| [2] |
K.M. Engle, T.S. Mei, M. Wasa, J.Q. Yu, Acc. Chem. Res. 45 (2012) 788-802. DOI:10.1021/ar200185g |
| [3] |
(a) J. He, M. Wasa, K.S.L. Chan, Q. Shao, J.Q. Yu, Chem. Rev. 117 (2017) 8754-8786; (b) J. Huang, Q. Gu, S.L. You, Chin. J. Org. Chem. 38 (2018) 51-61; (c) L. Xu, H. Xu, H. Lin, H. Dai, Chin. J. Org. Chem. 38 (2018) 1940-1948; (d) Q. Ren, B. Nie, Y. Zhang, J. Zhang, Chin. J. Org. Chem. 38 (2018) 2465-2490; (e) K. Zhao, L. Yang, J. Liu, C. Xia, Chin. J. Org. Chem. 38 (2018) 2833-2857; (f) Y.H. Liu, Y.N. Xia, B.F. Shi, Chin. J. Chem. 38 (2020) 635-662; (g) B. Ni, H. Cölfen, SmartMat 2 (2021) 17-32. |
| [4] |
(a) C. Shan, L. Zhu, L.B. Qu, R. Bai, Y. Lan, Chem. Soc. Rev. 47 (2018) 7552-7576; (b) M.M. Chen, L.Y. Shao, L.J. Lun, et al., Chin. Chem. Lett. 30 (2019) 702-706; (c) X.L. Han, P.P. Lin, Q. Li, Chin. Chem. Lett. 30 (2019) 1495-1502; (d) Y. Wu, B. Shi, Chin. J. Org. Chem. 40 (2020) 3517-3535. |
| [5] |
(a) F.L. Zhang, K. Hong, T.J. Li, H. Park, J.Q. Yu, Science 351 (2016) 252-256; (b) W. Liu, J. Zheng, Z. Liu, W. Hu, X. Wang, Y. Dang, ACS Catal. 8 (2018) 7698-7709. |
| [6] |
X. Jin, H. Xu, N. Zhao, R. Li, Y. Dang, Org. Lett. 22 (2020) 1464-1468. |
| [7] |
G. He, Y.S. Zhao, S.Y. Zhang, C.X. Lu, G. Chen, J. Am. Chem. Soc. 134 (2012) 3-6. DOI:10.1021/ja210660g |
| [8] |
E.T. Nadres, O. Daugulis, J. Am. Chem. Soc. 134 (2012) 7-10. DOI:10.1021/ja210959p |
| [9] |
(a) J.W. Xu, Z.Z. Zhang, W.H. Rao, B.F. Shi, J. Am. Chem. Soc. 138 (2016) 10750-10753; (b) B.B. Zhan, Y. Li, J.W. Xu, et al., Angew. Chem. Int. Ed. 57 (2018) 5858-5862. |
| [10] |
W. Cui, S.W. Chen, J.Q. Wu, et al., Org. Lett. 16 (2014) 4288-4291. DOI:10.1021/ol502011k |
| [11] |
(a) A. McNally, B. Haffemayer, B.S.L. Collins, M.J. Gaunt, Nature 510 (2014) 129-133; (b) J. Calleja, D. Pla, T.W. Gorman, V. Domingo, B. Haffemayer, M.J. Gaunt, Nat. Chem. 7 (2015) 1009-1016; (c) Y. Liu, H. Ge, Nat. Chem. 9 (2017) 26-32. |
| [12] |
H. Lin, X. Pan, A.L. Barsamian, T.M. Kamenecka, T.D. Bannister, ACS Catal 9 (2019) 4887-4891. DOI:10.1021/acscatal.8b04927 |
| [13] |
Y.F. Yang, G.J. Cheng, P. Liu, et al., J. Am. Chem. Soc. 136 (2014) 344-355. DOI:10.1021/ja410485g |
| [14] |
M. Anand, R.B. Sunoj, H.F. Schaefer, J. Am. Chem. Soc. 136 (2014) 5535-5538. DOI:10.1021/ja412770h |
| [15] |
W. Feng, T. Wang, D. Liu, X. Wang, Y. Dang, ACS Catal 9 (2019) 6672-6680. DOI:10.1021/acscatal.9b01412 |
| [16] |
D. Balcells, E. Clot, O. Eisenstein, Chem. Rev. 110 (2010) 749-823. DOI:10.1021/cr900315k |
| [17] |
D. Lapointe, K. Fagnou, Chem. Lett. 39 (2010) 1118-1126. DOI:10.1246/cl.2010.1118 |
| [18] |
D.L. Davies, S.A. Macgregor, C.L. McMullin, Chem. Rev. 117 (2017) 8649-8709. DOI:10.1021/acs.chemrev.6b00839 |
| [19] |
Y.F. Yang, X. Hong, J.Q. Yu, K.N. Houk, Acc. Chem. Res. 50 (2017) 2853-2860. DOI:10.1021/acs.accounts.7b00440 |
| [20] |
D.L. Davies, S.M.A. Donald, S. Macgregor, J. Am. Chem. Soc. 127 (2005) 13754-13755. DOI:10.1021/ja052047w |
| [21] |
A. Bondi, J. Phys. Chem. 68 (1964) 441-451. DOI:10.1021/j100785a001 |
| [22] |
J.E. Kickham, S.J. Loeb, Organometallics 14 (1995) 3584-3587. DOI:10.1021/om00007a071 |
| [23] |
P. Braunstein, C. Frison, N. Oberbeckmann-Winter, et al., Angew. Chem. Int. Ed. 43 (2004) 6120-6125. DOI:10.1002/anie.200461291 |
| [24] |
J. Vicente, A. Arcas, F. Julia-Hernandez, D. Bautista, Angew. Chem. Int. Ed. 50 (2011) 6896-6899. DOI:10.1002/anie.201102214 |
| [25] |
Y. Dang, S. Qu, J.W. Nelson, et al., J. Am. Chem. Soc. 137 (2015) 2006-2014. DOI:10.1021/ja512374g |
| [26] |
Y.F. Yang, X. Hong, G. Chen, J.Q. Yu, K.N. Houk, J. Am. Chem. Soc. 139 (2017) 8514-8521. DOI:10.1021/jacs.7b01801 |