 2021, Vol. 32
2021, Vol. 32 


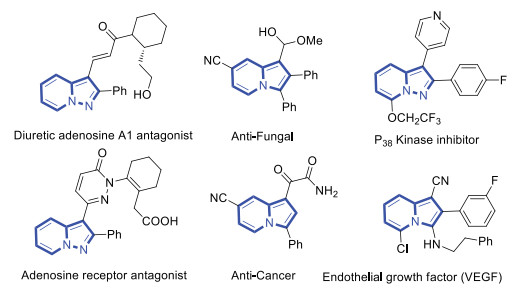
Nitriles are one of the most basic functional groups and are well represented in various functionalized materials and pharmaceuticals [1]. More than 50 nitrile-containing small-molecule drugs have been developed for various therapeutic indications according to the DrugBank database [2]. Therefore, development of robust and new synthetic strategies toward nitriles has garnered long-term interest in organic synthesis. Pyrazolo[1, 5-a]pyridines and indolizines are privileged structural motifs, occurring frequently in numerous natural products, pharmaceuticals, and biologically active molecules (Fig. 1) [3]. As a result, much effort has been directed toward exploring selective and efficient approaches to build these important heterocycle frameworks [4]. Representative routes to construct pyrazolo[1, 5-a]pyridines include [3 + 2] cycloaddition with N-aminopyridine derivatives [5], cyclization of N-iminopyridinium ylides [6], 2-substituted pyridines [7] and pyrazole rings [8] and functionalization of pyrazolo[1, 5-a]pyridine compounds [9]. On the other hand, the main strategies to produce indolizines are 1, 3-dipolar cycloaddition [10]b, transannulations of pyridotriazoles with alkynes [11], annulation of pyridine derivatives with unsaturated compounds [12], C−H functionalization reaction [13], and cyclization of N-substituted pyrroles [14]. Despite significant advances, the development of more concise and robust methods toward fused N-heterocycles is of great importance.

|
Download:
|
| Fig. 1. Examples of biologically active pyrazolo[1, 5-a]pyridines and indolizines. | |
Among ring-forming reactions, transition-metal-catalyzed oxidative annulation represents a straightforward strategy by enabling highly regioselective heterocycle syntheses [15]. Molecular oxygen is an ideal oxidant in oxygenation because of its abundance and environmental friendliness [16]. Furthermore, divergent synthesis that allows the rapid access to structurally diverse molecules from an identical substrate is highly desirable [17]. Very recently, Adimurthy group reported a copper-mediated annulation reaction of pyridyl esters and benzonitriles to afford pyrazolo[1, 5-a]pyridine-3-carboxylates. However, nitrile-substituted pyrazolo[1, 5-a]pyridine cannot be made by this method [7a]. Herein, we present the first divergent syntheses of nitrile-substituted pyrazolo[1, 5-a]pyridines and indolizines via palladium-catalyzed oxidative formal [4 + 1] annulation of pyridine-substituted acrylonitriles (Scheme 1).

|
Download:
|
| Scheme 1. Divergent synthesis of nitrile-containing pyrazolo[1, 5-a]pyridines and indolizines. | |
We commenced our investigation with acrylonitrile 1a and urea 2a under the Pd(OAc)2/Cu(NO3)2/O2 system at 120 ℃ for 4 h (Table 1, entry 1). Pyrazolo[1, 5-a]pyridine-3-carbonitrile 3a was isolated in 68% yield and the main byproduct was benzonitrile, which may be formed from the oxidative cleavage of the double bond of 1a. Then other common copper salts were examined in the reaction. Among the additive screened, Cu(OAc)2 exhibited the high efficiency and afforded the product in 76% yield (Table 1, entries 2–5). Subsequently, several solvents were tested, and other solvents were disadvantageous to this transformation (Table 1, entries 6–8). The control experiments revealed that no annulation products were formed without catalyst, additive and molecular oxygen, respectively (Table 1, entries 9–11). When NH4OH and NH4Cl were used as nitrogen source, 3a was furnished in lower yields compared with urea (Table 1, entries 12 and 13). It was unfavorable when the reaction time was shortened to 3 h (Table 1, entries 14 and 15). The reaction was conducted with Cu(OAc)2 in catalytic amount and the yield decreased (Table 1, entry 16). After the study of reaction conditions, we found that indolizine-1-carbonitrile 4a was achieved absolutely for switching the reaction conditions to Pd(OAc)2/FeCl3/O2 system (Table 1, entry 17). The same result was observed in the absence of urea, which implied that 2a did not participate in the reaction (Table 1, entry 18). Among the iron salts explored, FeBr3 was clearly the best choice (Table 1, entries 19–21). The yield was further increased to 91% when the reaction time was extended to 24 h (Table 1, entries 22 and 23). After a few attempts, the optimal conditions to pyrazolo[1, 5-a]pyridine-3-carbonitrile 3a is Cu(OAc)2 with urea and that to indolizine-1-carbonitrile 4a is FeBr3 for 24 h.
|
|
Table 1 Optimization of the reaction parameters for the switchable formation of products.a |

{kind=link}
{kind=link}
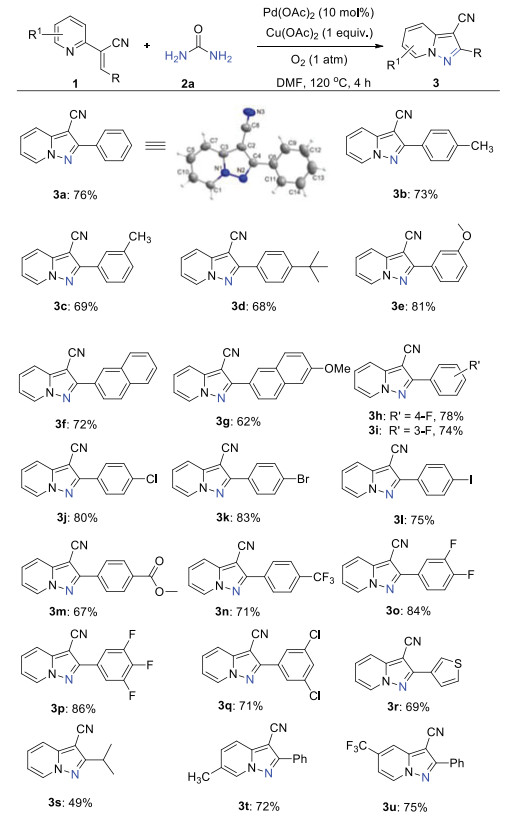
With optimized reaction conditions available, the substrate scope toward pyrazolo[1, 5-a]pyridine-3-carbonitriles 3 was first examined as shown in Scheme 2. In general, the annulation with pyridine-substituted acrylonitriles 1 bearing electron-donating groups and withdrawing groups in the aryl ring reacted well to render the desired products in moderate to good yields (3b-3q). The substrate containing tertiary butyl group delivered the product in 68% yield (3d). The reaction of the fused ring system also yielded the products in good yields (3f-3g). Importantly, halide substituents such as F, Cl, Br and I were well-tolerated. In particular, the heterocycles with aryl bromide and iodide could be further functionalized to produce more complicated structures (3h-3l). It is well known that fluorine-containing compounds have wide applications in medicinal chemistry and material science [18]. Interestingly, trifluoromethyl, difluoro and trifluoro-substituted substrates proceeded efficiently with urea to form the corresponding products in high yields (3n-3p). Fortunately, the heteroaryl substituted acrylonitrile could be achieved in 69% yield (3r). To our delight, the alkyl substituted substrate could generate the product 3s in moderate yield. Moreover, the substrates with substituents on the pyridine ring were also investigated and the products were achieved successfully (3t-3u). The structure of 3a was confirmed by X-ray crystal analysis (CCDC: 2050012). These results implied that this oxidative annulation can be effective to elaborate the pyrazolo[1, 5-a]pyridine-3-carbonitrile library.

|
Download:
|
| Scheme 2. Substrate scope of pyrazolo[1, 5-a]pyridine-3-carbonitriles. Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol), Pd(OAc)2 (10 mol%), and Cu(OAc)2 (1.0 equiv.) with DMF (1.0 mL) at 120 ℃ for 4 h in oxygen atmosphere, isolated yield. | |
{kind=link}
Next, the substrate scope of indolizines 4 was studied under the Pd(OAc)2/FeBr3/O2 system. As illustrated in Scheme 3, the palladium-catalyzed formal [4 + 1] annulation reactions were proven effective on syntheses of 2, 3-disubstituted indolizine-1-carbonitriles with good functional group tolerance (4a-4s). Initially, the phenyl ring moiety was investigated under the standard conditions. The bulky tert-butyl group and strong electron-donating groups were both accommodated in this transformation (4d-4f). Noteworthily, dimethoxy-substituted substrate smoothly worked and afforded 4g in 73% yield. Apart from phenyl ring, this reaction was also applicable for naphthyl group, and yielded the corresponding 4h and 4i in good yields. The reaction was compatible with a wide range of electron-deficient substituents, such as fluoro-, chloro-, bromo-, iodo-, ester- and trifluoromethyl groups (4j-4o). It should be pointed out that C-I bond is often incompatible with palladium catalytic system. The challenging substrate, trifluoro-substituted acrylonitrile also underwent homodifunctionalization reaction to give 4p product in 73% yield. In addition, the heterocycle substrate took place excellently and transferred into the desired product in satisfactory yield (4r). Unfortunately, when the alkyl species was subjected to this reaction, negative result was obtained. The structure of 4a was further confirmed by X-ray crystal diffraction measurement (CCDC: 2050013). Significantly, the reaction could be carried out on large-scale synthesis and provided the product 4a in 82% yield (0.36 g).

|
Download:
|
| Scheme 3. Substrate scope of indolizine-1-carbonitriles. Reaction conditions: 1 (0.3 mmol), Pd(OAc)2 (10 mol%), and FeBr3 (50 mol%) with DMF (1.0 mL) at 120 ℃ for 24 h in oxygen atmosphere; Isolated yield. a3 mmol scale of the reaction. | |
{kind=link}
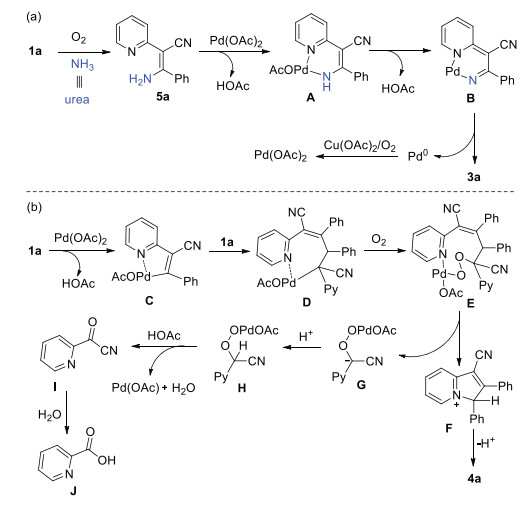
To gain insight into the reaction mechanism, some control experiments were performed (Scheme 4). Radical-trapping experiments were carried out in the presence of 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) under the standard conditions, and the yields only reduced slightly, which could exclude the radical mechanism (Scheme 4a). When 1a was subjected to the reaction with ammonia solution under oxygen atmosphere, enamine intermediate 5a [19] was obtained in good yield (Scheme 4b). To our delight, 5a could be transformed into 3a product under the standard conditions (Scheme 4c). These results inferred that enamine intermediate was involved in the reaction. Considering the indolizine products might be formed from the annulation of pyridine-substituted acrylonitriles with carbon surrogates, benzaldehyde, benzyl alcohol, toluene, or benzylamine was independently charged into the reaction mixture. However, only homodifunctionalization product 4b was produced (Scheme 4d). The experiment of mixing starting materials with different aryl groups was investigated briefly and resulted in a statistical distribution of homodifunctionalization products and cross-linked products were formed (Scheme 4e, GC ratio).

|
Download:
|
| Scheme 4. Control experiments. | |
{kind=link}
Based on the aforementioned experimental results and the literature precedents, a plausible mechanism toward pyrazolo[1, 5-a]pyridines was proposed (Scheme 5a). Firstly, the intermediate 5a was produced via the sequential Michael addition and oxidation process. Coordination of the enamine 5a with PdII generated intermediate A, affording intermediate B probably via cascade 1, 5-hydride transfer and ligand exchange with the release of HOAc. Subsequently, reductive elimination of B furnished the target product 3a and active Pd0 species. Finally, PdII was regenerated by the oxidation of CuII and oxygen [20].

|
Download:
|
| Scheme 5. Possible mechanism toward pyrazolo[1, 5-a]pyridines and indolizines. | |
{kind=link}
Although the exact reaction pathway remains elusive, a tentative reaction mechanism for indolizine formation was depicted (Scheme 5b). Initially, electrophilic attack of PdII on the pyridine-substituted acrylonitrile 1a formed intermediate C, which underwent a Heck cross-coupling with 1a to generate intermediate D and then attacked by O2 forming intermediate E. Subsequently, the intermediate F was formed by the nucleophilic attack of the pyridine nitrogen with the cleavage of the C-C bond, and the intermediate G was afforded at the same time. Finally, the 4a product was yielded by the deprotonation process. On the other hand, the reactive acyl cyanide I was achieved via the 1, 2-elimination of intermediate H, which was hydrolyzed to picolinic acid J [21]. The picolinic acid byproduct can be detected in GC–MS spectrometer. It is likely that FeBr3 as Lewis acid promotes the annulation process.
In summary, we have developed a highly efficient and selective syntheses of nitrile-substituted pyrazolo[1, 5-a]pyridines and indolizines from pyridine-substituted acrylonitriles via the palladium catalysis. The reaction involves the cleavage of C-N/C-C bonds and the formation of new C-C/C-N/N-N bonds. The advantages of this transformation include readily accessible substrates, cheap and safe urea as nitrogen source, divergent synthesis, controllable selectivity, oxygen participated reaction, good functional group tolerance, etc. In addition, nitrile-substituted fused N-heterocycles have potential applications in synthetic and pharmaceutical chemistry. Further synthetic utilization and mechanistic studies are currently ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for financial support from the Natural Science Foundation of Jiangxi Provincial Education Department (Nos. GJJ201422, GJJ190754, GJJ190776) and Science and Technology Project of Ganzhou City. We acknowledge the Analytical & Testing Center of Beijing Normal University for the high-resolution mass spectrometry analyses.
| [1] |
(a) G. Yan, Y. Zhang, J. Wang, Adv. Synth. Catal. 359 (2017) 4068–4105; (b) Y. Ping, Q. Ding, Y. Peng, ACS Catal. 6 (2016) 5989–6005; (c) J. Kim, H.J. Kim, S. Chang, Angew. Chem., Int. Ed. 51 (2012) 11948–11959; (d) P. Anbarasan, T. Schareina, M. Beller, Chem. Soc. Rev. 40 (2011) 5049–5067; (e) G.P. Ellis, T.M. Romney-Alexander, Chem. Rev. 87 (1987) 779–794. |
| [2] |
Y. Wang, Y. Du, N. Huang, Future Med. Chem. 10 (2018) 2713-2727. |
| [3] |
(a) J.T.M. Correia, B. List, F. Coelho, Angew. Chem. Int. Ed. 56 (2017) 7967–7970; (b) D. Chandra Mohan, C. Ravi, V. Pappula, et al., J. Org. Chem. 80 (2015) 6846–6855; (c) V. Sharma, V. Kumar, Med. Chem. Res. 23 (2014) 3593–3606; (d) J.D. Kendall, Curr. Org. Chem. 15 (2011) 2481–2518. |
| [4] |
(a) B. Nie, W. Wu, Y. Zhang, et al., Org. Chem. Front. 7 (2020) 3067–3099; (b) B. Sadowski, J. Klajn, D.T. Gryko, Org. Biomol. Chem. 14 (2016) 7804–7828; (c) G.S. Singh, E.E. Mmatli, Eur. J. Med. Chem. 46 (2011) 5237–5257. |
| [5] |
(a) V.A. Motornov, A.A. Tabolin, Y.V. Nelyubina, et al., Org. Biomol. Chem. 18 (2020) 1436–1448; (b) Q. Huang, D. He, J. Han, et al., Synthesis 50 (2018) 3731–3737; (c) X. Yu, S. Ye, J. Wu, Adv. Synth. Catal. 352 (2010) 2050–2056; (d) Z. Chen, J. Wu, Org. Lett. 12 (2010) 4856–4859; (e) S. Patnaik, H.C. Dietz, W. Zheng, et al., J. Org. Chem. 74 (2009) 8870–8873. |
| [6] |
J.J. Mousseau, J.A. Bull, C.L. Ladd, et al., J. Org. Chem. 76 (2011) 8243–8261, and references cited therein
|
| [7] |
(a) D.C. Mohan, C. Ravi, S.N. Rao, et al., Org. Biomol. Chem. 13 (2015) 3556–3560, and references cited therein; (b) K.L. Stevens, D.K. Jung, M.J. Alberti, et al., Org. Lett. 7 (2005) 4753–4756. |
| [8] |
O. Obulesu, V. Murugesh, B. Harish, et al., J. Org. Chem. 83 (2018) 6454-6465. DOI:10.1021/acs.joc.8b00746 |
| [9] |
(a) G. Yan, Y. Zhang, J. Wang, Adv. Synth. Catal. 359 (2017) 4068–4105; (b) K. Alam, S.M. Kim, D.J. Kim, et al., Adv. Synth. Catal. 358 (2016) 2661–2670. |
| [10] |
(a) W. Wang, J. Han, J. Sun, et al., J. Org. Chem. 82 (2017) 2835–2842; (b) B. Shen, B. Li, B. Wang, Org. Lett. 18 (2016) 2816–2819; (c) J. Brioche, C. Meyer, J. Cossy, Org. Lett. 17 (2015) 2800–2803. |
| [11] |
(a) Y. Shi, V. Gevorgyan, Chem. Commun. 51 (2015) 17166–17169; (b) V. Helan, A.V. Gulevich, V. Gevorgyan, Chem. Sci. 6 (2015) 1928–1931; (c) S. Chuprakov, F.W. Hwang, V. Gevorgyan, Angew. Chem. Int. Ed 46 (2007) 4757–4759; (d) S. Chuprakov, V. Gevorgyan, Org. Lett. 9 (2007) 4463–4466. |
| [12] |
(a) Z. Chen, P. Liang, F. Xu, et al., J. Org. Chem. 84 (2019) 12639–12647; (b) X. Wu, P. Zhao, X. Geng, et al., Org. Lett. 19 (2017) 3319–3322; (c) Y. Liu, Y. Yu, Y. Fu, et al., Org. Chem. Front. 4 (2017) 2119–2123; (d) H. Li, X. Li, Y. Yu, et al., Org. Lett. 19 (2017) 2010–2013; (e) S. Tang, K. Liu, Y. Long, et al., Chem. Commun. 51 (2015) 8769–8772; (f) S. Tang, K. Liu, Y. Long, et al., Org. Lett. 17 (2015) 2404–2407; (g) R.R. Liu, J.J. Hong, C.J. Lu, et al., Org. Lett. 17 (2015) 3050–3053; (h) X. Wang, S.Y. Li, Y.M. Pan, et al., Org. Lett. 16 (2014) 580–583. |
| [13] |
(a) J.T. Menezes Correia, B. List, F. Coelho, Angew. Chem., Int. Ed 56 (2017) 7967–7970; (b) M.F.Z.J. Amaral, A.A. Baumgartner, R. Vessecchi, et al., Org. Lett. 17 (2015) 238–241; (c) Q. Wu, D. Zhao, X. Qin, et al., Chem. Commun. 47 (2011) 9188–9190; (d) Y. Yang, K. Cheng, Y. Zhang, Org. Lett. 11 (2009) 5606–5609; (e) J.B. Xia, S.L. You, Org. Lett. 11 (2009) 1187–1190. |
| [14] |
(a) X. Li, X. Xie, Y. Liu, J. Org. Chem. 81 (2016) 3688–3699; (b) W. Hao, H. Wang, Q. Ye, et al., Org. Lett. 17 (2015) 5674–5677; (c) J.H. Lee, I. Kim, J. Org. Chem. 78 (2013) 1283–1288; (d) M. Kucukdisli, T. Opatz, J. Org. Chem. 78 (2013) 6670–6676. |
| [15] |
(a) D. Wang, A.B. Weinstein, P.B. White, et al., Chem. Rev. 118 (2018) 2636–2679; (b) A.V. Gulevich, A.S. Dudnik, N. Chernyak, et al., Chem. Rev. 113 (2013) 3084–3213; (c) J. Wencel-Delord, T. Dröge, F. Liu, et al., Chem. Soc. Rev. 40 (2011) 4740–4761; (d) T.W. Lyons, M.S. Sanford, Chem. Rev. 110 (2010) 1147–1169; (e) E.M. Beccalli, G. Broggini, M. Martinelli, et al., Chem. Rev. 107 (2007) 5318–5365. |
| [16] |
W. Wu, H. Jiang, Acc. Chem. Res. 45 (2012) 1736-1748. DOI:10.1021/ar3000508 |
| [17] |
(a) F.G. Kuruvilla, A.F. Shamji, S.M. Sternson, et al., Nature 416 (2002) 653–657; (b) S.L. Schreiber, Science 287 (2000) 1964–1969. |
| [18] |
(a) C. Alonso, E. Martínez de Marigorta, G. Rubiales, et al., Chem. Rev. 115 (2015) 1847–1935; (b) J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432–2506. |
| [19] |
M. Wang, J. Hou, W. Yu, et al., J. Org. Chem. 83 (2018) 14954-14961. DOI:10.1021/acs.joc.8b02022 |
| [20] |
S.S. Stahl, Angew. Chem. Int. Ed. 43 (2004) 3400-3420. DOI:10.1002/anie.200300630 |
| [21] |
X. Xu, B. Li, Y. Zhao, et al., Org. Chem. Front. 4 (2017) 331-334. DOI:10.1039/C6QO00635C |