 2021, Vol. 32
2021, Vol. 32 


b School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China;
c State Key Laboratory of Modern Optical Instrumentation, College of Optical Science and Engineering, Hangzhou 310027, China
Fluorescence is the cornerstone of numerous cut-edge biological and medical technologies [1-6], e.g., fluorescence in situ hybridization (FISH) [7], cell imaging [8], DNA sequencing [9], flow cytometry [10], super-resolution imaging [11], and fluorescence guided-surgery [12]. Fluorescent materials of different chemical natures were reported, e.g., organic fluorophores [13], inorganic quantum dots [14], carbon materials [15], lanthanide complexes, up-converting inorganic materials [16], polymeric emitters [17], and biological fluorescent proteins [18]. These materials have unique merits and limitations and mutually complementary. For in vitro or in vivo applications, organic fluorophores are particularly suitable sought after due to their biocompatibility, spectral versatility, and facile derivatization for functionality [19].
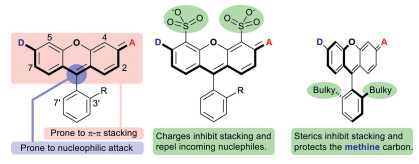
Rhodamine is a premium class of organic fluorophores. They are conveniently synthesized from inexpensive starting materials, structurally rigid, and therefore very bright. Their spectral properties can be readily fine-tuned in the range of ca. 490 of Rh110 [20] to 590 nm of Texas Red [21]. Additionally, they can be engineered to exhibit high resistance toward nucleophilic attack or aggregation by sulfonation at C-4/C-5 of its xanthene core, or installation of bulky groups at C3′/C7′ of the bottom benzene ring (Fig. 1) [22]. Recent years have witnessed a surge for rhodamines analogs absorbing/emitting beyond 600 nm, by replacing the central oxygen atom of the xanthene core into a less electron-donating moiety than oxygen. The earliest endeavor is the class of dyes commonly referred to as carbo-rhodamines [23], which has an sp3 hybridized carbon moiety as the bridging group. They exhibit a reduced tendency to aggregate, but an enhanced susceptibility toward nucleophilic attack. While the nucleophilic attack by thiols or hydride is reversible and can be harnessed for good in localization microscopy, the attack by oxidative nucleophiles such as peroxynitrite, hypochloride, superoxide, peroxide could lead to permanent destruction of the rhodamine scaffold and place a restriction over the scope of applicable scenarios. Unfortunately, these above methods to promote the chemostability of rhodamine dyes are not applicable to carbo-rhodamines because the steric hindrance of the central disubstituted methylene unit renders the C4/C5 and C3′/C7′ positions inaccessible for chemical modifications.

|
Download:
|
| Fig. 1. The generic structure of rhodamine dyes, their limitations, and remedies. | |
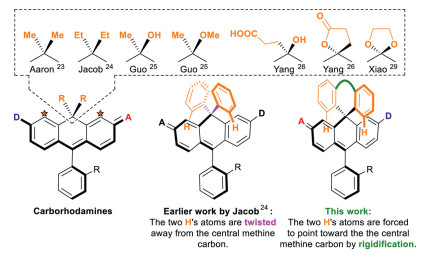
The use of bulkier chemical groups other than methyl is an obvious solution. For example, Jacob et al. prepared diethyl and diphenyl substituted carbo-rhodamines [24]. Recently, Guo et al. recently reported a new synthesis of carbo-rhodamines allowing replacement of one of the two methyl groups into a hydroxyl or methoxyl, or potentially other alkoxyl groups [25]. We prepared two facilely substituted carbo-rhodamines, with a combination of carboxyethyl/hydroxyl groups, and a dihydrofuran-2(3H)-one, respectively [26]. Xiao et al. reported carbo-rhodamines with a 1,3-dioxolane bridge [27]. Yet, these groups are pointing away from the electrophilic central methine carbon and does not impart sufficient steric protection toward nucleophilic attack. Having the two alkyl groups replaced by two bulkier phenyl groups does not address this difficulty either because the two phenyl rings are rotatable and they tend to adopt a geometry, in which the two peripheral hydrogen atoms are also twisted away from the central methine carbon and not provide much steric protection over the central methine carbon. We envisage that the steric protection of the two phenyl group could be greatly enhanced if they are tethered together. This way, the two hydrogen atoms of the phenyl rings, highlighted in orange colour, are forced to point down toward the central methine carbon (Fig. 2). Following this line of research, our first attempt is a rhodamine with a benzophenone bridge. They exhibit the desired high chemical stability, and however is still difficult for further functionalization due to its electron deficient nature.

|
Download:
|
| Fig. 2. The generic structures of existing carbo-rhodamines and the main idea of this work to develop novel highly stable xanthenoid dyes. | |
In this work, it is our goal to design and synthesize new carbo-rhodamine analogs, which not only exhibit high resistance toward aggregation or nucleophilic attack, but also allow convenient further modification. Herein, we report the design, synthesis, spectral studies and proof-of-concept applications of fluoreno-xanthene (10) and xantheno-xanthene (16a and 16b), i.e. xanthene dyes with a "fluorene" or a "xanthene" bridging group, which fulfils the requirements discussed previously (Scheme 1).

|
Download:
|
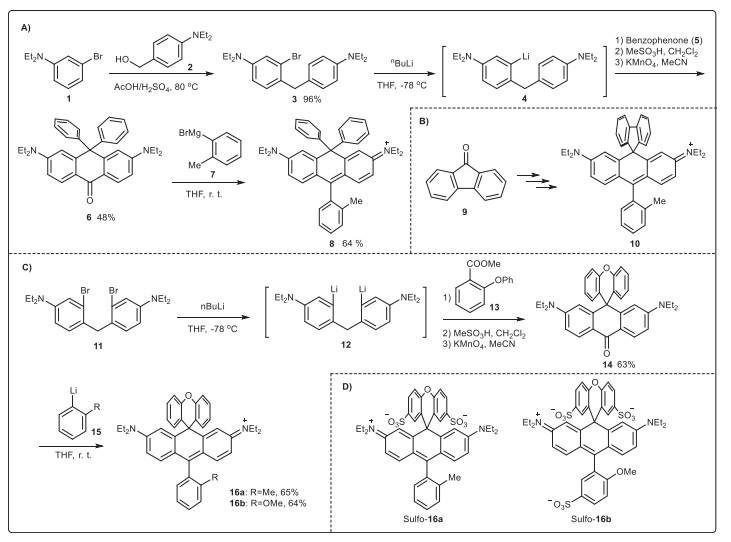
| Scheme 1. Synthesis of various new carbo-rhodamines (10, 16a, 16b, Sulfo-16a, Sulfo-16b). | |
We devised a new synthesis for carbo-rhodamine dyes, and the feasibility was first exemplified by synthezing a bis-phenyl substituted carbo-rhodamine (8). 3-Bromo-N, N-diethylaniline (1) was prepared via alkylation of 3-bromoaniline with EtI in a nearly quantitative yield. Acid-catalysed condensation of 1 with (4-(diethylamino)phenyl)methanol (2) at 80 ℃ for 3 in a 96% yield. Compound 3 was treated with nBuLi (1.2 equiv.) at -78 ℃ for 15 min to prepare the corresponding lithium reagent in situ via halogen-lithium exchange before benzophenone 5 (1.0 equiv. in THF) was added to furnish (5-diethylamino-2-(4-diethylaminobenzyl)phenyl)diphenylmethanol, which was refluxed with MeSO3H in CH2Cl2 for 2 h and then oxidized to 3,6-bis(diethylamino)-10,10-diphenylanthracen-9(10H)-one (6) by KMnO4 in an overall 48% yield. Compound 6 reacted with Grignard reagent (7, 2 equiv.) at 45 ℃ for 5 h to produce 8 in a 65% yield. Dye 10 was synthesized similarly with fluorenone (9) instead of benzophenone (5) in an overall 37% yield. Surprisingly, the preparation of 16a and 16b from xanthenone was not successful. So, we proposed an alternative synthesis starting from 4,4′-methylenebis(3-bromo-N, N-diethylaniline) (11), whose synthesis was reported during the first synthesis of silicon rhodamine by Xiao and Qian et al. [28, 29]. Compound 11 was treated with nBuLi (2.4 equiv.) at -78 ℃ for 15 min to prepare the corresponding lithium reagent in situ via halogen-lithium exchange before methyl 2-phenoxybenzoate 13 (1.0 equiv.) was added to furnish 2,7-bis(diethylamino)-9-(2-phenoxyphenyl)-9,10-dihydroanthr-acen-9-ol, which was oxidized by KMnO4 before refluxing with MeSO3H in CH2Cl2 for 2 h to produce the desired 2,7-bis(diethylamino)-10H-spiro[anthracene-9,9′-xanthen]-10-one (14) in a 63% yield. Compound 14 was further treated with Grignard reagent (15a and 15b, 2 equiv.) at 45 ℃ for 5 h to produce 16a and 16b in good yields. The single-crystal structure of 16b was obtained (Fig. S1 in Supporting information). The upper diphenyl ether plane is perpendicular to the fluorochromic xanthene structure.
We tested the feasibility of 16a and 16b for functionalization, i.e., sulfonation, which is a valued substitution for dyes because it improves water solubility, photo/chemo stability, and resistance toward aggregation simultaneously. Compound 10 was dissolved in concentrated H2SO4 in an ice bath and stirred for 24 h, and no desired sulfonated derivatives were observed. Presumably, the biphenyl moiety is not sufficiently electron-rich. We then tested 16a and 16b. Under the same condition, the diphenyl ether moiety of 16a was smoothly bis-sulfonated to Sulfo-19a in a 64% yield. Sulfonation of 16b produced a tris-sulfonated product (Sulfo-16b) in a 64% yield.
Dyes 16b and Sulfo-16b were selected for further spectral and imaging studies. Dye 16b has an absorption band typical of a xanthene dye, including a major peak with a maximum at ca. 640 nm in such solvents as CH2Cl2/DMSO/MeOH/phosphate buffer with their molar absorptivities ranging of (1.0-1.3) × 105 L mol-1 cm-1 and a shoulder peak. The emission band of 16b is a good mirror image of their absorption band, including a major peak at ca. 660 nm in different solvents and a tailing shoulder band. It is particularly noteworthy that the fluorescence quantum yields in these solvents are very high, ranging from 0.91 in CH2Cl2, 0.66 in DMSO, 0.69 in MeOH, to 0.46 in phosphate buffer. The maximum absorption and emission wavelengths of Sulfo-16b exhibit a slight bathchromic shift of ca. 5-10 nm compared to 16b in the same solvents. Comparatively the molar absorptivities and the fluorescence quantum yields witnessed a drop. Nevertheless, Sulfo-16b is still a bright dye with a molar absorptivity of 81,000 L mol-1 cm-1 and quantum yield of 0.38 in phosphate buffer. We note that the spectral properties of Sulfo-16b are essentially the same to those of a classic Cy5 dye (Sulfo-Cy5). The molar absorptivity of Sulfo-Cy5 is high at 234,000 L mol-1 cm-1 and the fluorescence yield is lower at 0.27. (Table 1 and Fig. S2 in Supporting information).
|
|
Table 1 The spectral properties of 16b, Sulfo-16b and Sulfo-Cy5 in different solvents.a |

{kind=link}
{kind=link}
{kind=link}
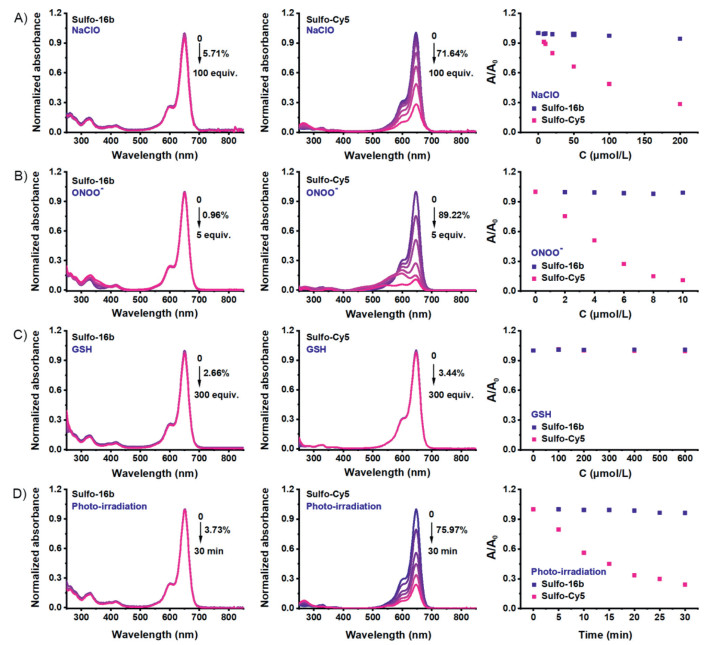
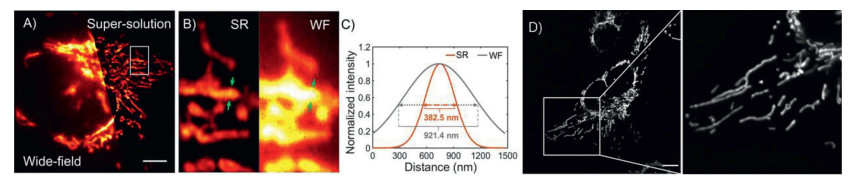
Therefore, 16b and Sulfo-16b can potentially replace Cy5 dyes for practical applications. Cy5 type fluorophores are known for their poor stabilities. Their floppy polymethine backbone is prone to nucleophilic attack by thiolates or reactive oxygen species, including hypochlorite, peroxynitrite, etc. Therefore, chemo-/photo-stabilities of Sulfo-16b were studied with Sulfo-Cy5 as a reference. An addition of hypochlorite (up to 100 equiv., Fig. 3A) or peroxynitrite (up to 5 equiv., Fig. 3B) caused a dose-dependent decrease of the absorbance of Sulfo-Cy5 at 648 nm, verifying it poor chemo-stabilities toward these reactive oxygen species. Under the same condition, Sulfo-16b remained unchanged, suggesting that Sulfo-16b is chemically stable even in a large excess of these oxidative species. The stabilities of Sulfo-Cy5 and Sulfo-16b toward GSH (up to 300 equiv., Fig. 3C) were checked and both remained stable. Lastly, the photo-stability of Sulfo-16b and Sulfo-Cy5 were compared by irradiation of their solution in neutral phosphate buffer with a halogen lamp (Fig. 3D). In 30 min, the absorbance of Sulfo-Cy5 decreased by 80%, while the absorbance of Sulfo-16b again remains unchanged. Absorption/emission maxima in the deep-red region, high chemo-/photo-stabilities, and bright fluorescence render 16b and its hydrophilic Sulfo-16b value fluorophores for bio-imaging. Their potentials for imaging were showcased with 16b in live U2OS cells via three different microscopic methods, i.e., epi-fluorescence, structured illumination (SIM), and stimulated emission depletion (STED). Cells were incubated with compound 16b (1.2 µmol/L) for 10 min, rinsed with PBS three times, and cell images were acquired. Bright images of filamentous structures, presumably mitochondria, were observed by all three methods (Fig. 4 for wide-field/SIM and STED). Cells did not exhibit structural abnormalities, and the fluorescence did not exhibit noticeable bleaching during the course of imaging. Though preliminary, these imaging results are sufficient to verify that 16b and Sulfo-16b are feasible for cell-based imaging applications.

|
Download:
|
| Fig. 3. Normalized absorption spectra over time for the solutions of Sulfo-16b and Sulfo-Cy5 in PBS (pH 7.4) with the addition of different equivalents of NaClO (A), ONOO- (B), GSH (C), and upon irradiation with a halogen lamp (300 W) (D). | |
{kind=link}

|
Download:
|
| Fig. 4. (A) Wide-field and reconstructed SIM images of cell mitochondria in U2OS cells with 16b. (B) Further magnified region of interest (ROI). (C) The intensity profiles of mitochondria in super-resolution SIM image and wide-field image. Exc: 640 nm. Scale bar: 5 µm. (D) STED imaging of U2OS cells incubated with 16b and the magnified image of the region of interest (ROI). Exc: 640 nm; Depletion lasers: 775 nm; Scale bar: 10 µm. | |
{kind=link}
Often, xanthene dyes are installed with various functionalities, e.g., benzyl bromide, amino, carboxyl, or activated carboxyl. We showcase that this chemistry is also applicable to the novel carbo-rhodamines reported herein. The benzephenone 14 was reacted with 2-methoxymethylphenyl lithium to prepare dye 17, which was further treated with BBr3 to furnish the dye (18) with a benzyl bromide, which is a good reaction partner for various biological thiols. Then, by reacting 19 with aminothiol ethanol or 3-mercaptopropanoic acid, respectively, dyes with a primary amino group or a carboxyl group were prepared in excellent yields. If necessary, the carboxyl group of 20 can be conveniently activated into a succinimidyl ester (21) for conjugation with an amino group (Scheme S1 in Supporting information).
Carbo-rhodamine dyes are small-molecule fluorophores found wide applications. Yet, they have such limitations as susceptibility toward nucleophilic attack at the central methine carbon, and lack of ample positions for chemical derivatization since the C4/C5 positions are not available for reaction due to sterics of the methylene bridge. At the methylene bridge, we proposed to install bulky groups capable of sterically shielding the methine carbon to impart high stability. Following this idea, fluoreno-xanthene and xantheno-xanthene dyes were prepared. Xantheno-xanthene dyes are particularly favorably for practical applications because their long-wavelength absorption/emission maxima, high chemo-/photo-stabilities, bright fluorescence, and viability for sulfonation. We further showcased that they have potentials for various microscopic methods, including the very demanding STED super-resolution imaging. Lastly, these scaffolds are readily derivatized with chemical handles for bioconjugation. The general principles presented in this manuscript can be apparently translated to other xanthenoids dyes with a silicon-or phosphorous-based bridging group.
Declaration of competing interestWe declare no conflict-of-interests of any kind.
AcknowledgmentsThe work is financially supported by the National Natural Science Foundation of China (Nos. 21822805, 21908065, 22078098), the Science and Technology Commission of Shanghai Municipality for the Shanghai International Cooperation Program (No. 18430711000), the China Postdoctoral Science Foundation (Nos. 2019M651427, 2020T130197).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.04.041.
| [1] |
Z. Cheng, T. Zhang, W. Wang, et al., Chin. Chem. Lett. 32 (2021) 1580-1585. DOI:10.1016/j.cclet.2021.02.017 |
| [2] |
D. Li, W. Chen, S.H. Liu, et al., Chin. Chem. Lett. 31 (2020) 2891-2896. DOI:10.1016/j.cclet.2020.02.047 |
| [3] |
K. Wang, W. Ma, Y. Xu, et al., Chin. Chem. Lett. 31 (2020) 3149-3152. DOI:10.1016/j.cclet.2020.08.039 |
| [4] |
C. Yan, L. Shi, Z. Guo, W. Zhu, Chin. Chem. Lett. 30 (2019) 1849-1855. DOI:10.1016/j.cclet.2019.08.038 |
| [5] |
Y. Chen, L. Li, W. Chen, et al., Chin. Chem. Lett. 30 (2019) 1353-1360. DOI:10.1016/j.cclet.2019.02.003 |
| [6] |
S. Long, Q. Qiao, L. Miao, Z. Xu, Chin. Chem. Lett. 30 (2019) 573-576. DOI:10.1016/j.cclet.2018.11.031 |
| [7] |
D. Huber, L.V. von Voithenberg, G.V. Kaigala, Micro Nano Eng. 1 (2018) 15-24. DOI:10.1016/j.mne.2018.10.006 |
| [8] |
L. Kacenauskaite, N. Bisballe, R. Mucci, et al., J. Am. Chem. Soc. 143 (2021) 1377-1385. DOI:10.1021/jacs.0c10457 |
| [9] |
C.R.J.Y. Ju, C.W. Fullert, A.N. Glazer, R.A. Mathies, Proc. Natl. Acad. Sci. U. S. A. 92 (1995) 4347-4351. DOI:10.1073/pnas.92.10.4347 |
| [10] |
H. Mikami, M. Kawaguchi, C.J. Huang, et al., Nat. Commun. 11 (2020) 1162. DOI:10.1038/s41467-020-14929-2 |
| [11] |
Z. Ye, W. Yang, C. Wang, et al., J. Am. Chem. Soc. 141 (2019) 14491-14495. DOI:10.1021/jacs.9b04893 |
| [12] |
R.Q. Yang, K.L. Lou, P.Y. Wang, et al., Small Methods (2021) 2001066. DOI:10.1002/smtd.202001066 |
| [13] |
Q. Zheng, M.F. Juette, S. Jockusch, et al., Chem. Soc. Rev. 43 (2014) 1044-1056. DOI:10.1039/C3CS60237K |
| [14] |
E. Scholl, L. Hanschke, L. Schweickert, et al., Nano Lett. 19 (2019) 2404-2410. DOI:10.1021/acs.nanolett.8b05132 |
| [15] |
Y. Huang, L. Xiao, T. An, et al., Small 13 (2017) 1700869. DOI:10.1002/smll.201700869 |
| [16] |
J. Peng, Y. Wang, J. Wang, et al., Biosens. Bioelectron. 28 (2011) 414-420. DOI:10.1016/j.bios.2011.07.057 |
| [17] |
K. Gardner, M. Aghajamali, S. Vagin, et al., Adv. Funct. Mater. 28 (2018) 1802759. DOI:10.1002/adfm.201802759 |
| [18] |
G.J. Kremers, S.G. Gilbert, P.J. Cranfill, et al., J. Cell Sci. 124 (2011) 157-160. DOI:10.1242/jcs.072744 |
| [19] |
J. Wiedenmann, F. Oswald, G.U. Nienhaus, IUBMB Life 61 (2009) 1029-1042. DOI:10.1002/iub.256 |
| [20] |
S.S. Chandran, K.A. Dickson, R.T. Raine, J. Am. Chem. Soc. 127 (2005) 1652-1653. DOI:10.1021/ja043736v |
| [21] |
R.H.J.A. Titus, S.O. Sharrow, D.M. Segal, J. Immunol. Methods 50 (1982) 193-204. DOI:10.1016/0022-1759(82)90225-3 |
| [22] |
Z. Lei, X. Li, Y. Li, et al., J. Org. Chem. 80 (2015) 11538-11543. DOI:10.1021/acs.joc.5b01746 |
| [23] |
C. Aaron, C.C. Barker, J. Chem. Soc. (1963) 2655-2662. DOI:10.1039/jr9630002655 |
| [24] |
J. Arden-Jacob, J. Frantzeskos, N.U. Kemnitzer, et al., Spectrochim. Acta A 57 (2001) 2271-2283. DOI:10.1016/S1386-1425(01)00476-0 |
| [25] |
X. Lv, T. Han, X. Yuan, et al., Analyst 146 (2021) 64-68. DOI:10.1039/D0AN01916J |
| [26] |
K. Xin, X. Li, Y. Guo, et al., CCS Chem. 2 (2020) 2307-2315. |
| [27] |
X. Zhang, L. Chen, Z. Huang, et al., Chem. Eur. J. 27 (2021) 3688-3693. DOI:10.1002/chem.202005296 |
| [28] |
M. Fu, Y. Xiao, X. Qian, et al., Chem. Commun. (2008) 1780-1782. |
| [29] |
Y. Xiao, X. Qian, Coord. Chem. Rev. 423 (2020) 213513. DOI:10.1016/j.ccr.2020.213513 |
| [30] |
R.B. Mujumdar, L.A. Ernst, S.R. Mujumdar, et al., Bioconjug. Chem. 4 (1993) 105-111. DOI:10.1021/bc00020a001 |