 2021, Vol. 32
2021, Vol. 32 


b Qian Xuesen Collaborative Research Center of Astrochemistry and Space Life Sciences, Ningbo University, Ningbo 315211, China;
c Department of Chemistry and Nano Science, Ewha Womans University, Seoul 03760, Korea
Oxidative activation of the inert C–H and C=C bond with high selectivity in benign environments is still a Holly Grail in organic synthesis [1, 2]. However, such oxidations are well accomplished in nature, mainly by heme [3] (e.g., cytochrome P450) and nonheme [4] (e.g., methane monooxygenase) enzymes. Uptaking electrons and protons from the protein environment, these enzymes form several iron-oxygen intermediates, such as the iron-superoxo (Fe+3–OO·) [5], the iron-peroxo (Fe+3–OO) [6], the iron-hydroperoxo (Fe+3–OOH) [7], and the high-valent iron-oxo (Fe+4/+5=O) reactive intermediates [8, 9], which are speculated as the active reaction intermediate to perform the robust oxidations. To directly obtain the putative high-valent iron-oxo intermediates, iodosylarenes (e.g., iodosylbenzene, PhIO) have been introduced into the enzymatic and biomimetic reaction systems. Metal-iodosylarene adducts (M-PhIO) are normally formed after the introduction of PhIO (Scheme 1a). These adducts are believed to be very unstable and decay automatically to the putative high-valent metal-oxo oxidants, which performs various kinds of oxidations. Such one-oxidant mechanism proposed by Groves and his coworkers has been widely accepted in the heme and nonheme chemistry, after combination of the oxygen-rebound mechanism [10] and the concept of two-state reactivity [11-13].

|
Download:
|
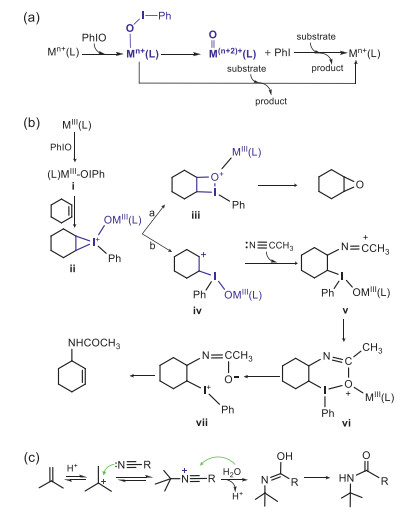
| Scheme 1. Schematic plots of (a) the multiple-oxidant mechanism, (b) the iodine(Ⅲ) chemistry proposed by Valentine, and (c) the traditional Ritter mechanism. | |
{kind=link}
However, Valentine and co-workers found in 1990 that olefin oxidation with iodosylbenzene could be significantly promoted by the redox-inert metal salts, such as Zn(OTf)3 and Al(OTf)3, which cannot form the high-valent metal-oxo oxidants (e.g., the Zn+4O and the Al+5O species) from the metal-iodosylarene precursors. Meanwhile, the products formed in the oxidation reactions mediated by Al(OTf)3 and by the redox-active iron salt Fe(OTf)3 are nearly identical, indicating that a common oxidant and a similar reaction mechanism work in these oxidations [14-16]. These unexpected results casted the one-oxidant mechanism into doubt. In 2002, Nam and his co-workers proposed the multiple-oxidant mechanism (Scheme 1a) in their chiral epoxidation with various iodosylarenes mediated by heme catalysts [17]. In this mechanism, both the metal-iodosylarene adducts and the high-valent metal-oxo complexes are the active oxidants to perform oxidations. Many experimental and theoretical studies have provided evidence to support the multiple-oxidant mechanism [18-24]. Thus, debates on the two dichotomous one-oxidant/multiple-oxidant mechanisms become prevalent and this controversy has been a hot topic in the biomimetic nonheme chemistry. The key point to solve this controversy is to unveil the elusive structure-reactivity relationship of the metal-iodosylarene adducts. In 2017, we revealed that the metal-iodosylarene adducts can generate two transient resonance valence bond structures, a high-valent metal-oxo species and a monomeric PhIO one, as a response to different substrate approaching orientations [21]. Very recently, an unusual hydride transfer/proton transfer (HT/PT) mechanism involving a carbonium intermediate was proposed in desaturation of 9, 10-dihydroantharacene (DHA) by a manganese-iodosylarene complex [18] and a cobalt-iodylarene complex [19]. The formation of an unusual carbonium intermediate via hydride transfer in the C–H bond activation inspires us to reinvestigate the mechanism proposed by Valentine (Scheme 1b) [14, 16] in the cyclohexene oxidation with PhIO catalyzed by inorganic metal salts (e.g., Fe(OTf)3). They proposed the metal-iodosylarene adducts M-PhIO (ⅰ in Scheme 1b), instead of the high-valent metal-oxo species as the oxidant. The oxidation reaction is initiated by an electrophilic addition of iodine(Ⅲ) to the C=C double bond to form a triangular C-C-I(Ⅴ) species ⅱ. Such triangular species is unstable and decays by two pathways: in path a, ⅱ decays to a quadrangular C-C-O-I(Ⅲ) species ⅲ, which subsequently converts to an epoxide, whereas in path b, ⅱ convers to a cyclohexane carbonium complex with an iodine(Ⅲ) species ⅳ. Such carbonium intermediate is ready to bind with an acetonitrile molecule via the well-known Ritter mechanism [25, 26] (Scheme 1c) to generate a carbonium amide ⅴ, which produces a carbonyl-amide-iodine(Ⅲ) complex ⅶ via a cyclic species ⅵ. The cyclohexene amide product forms by the decomplexation of PhI from ⅶ.
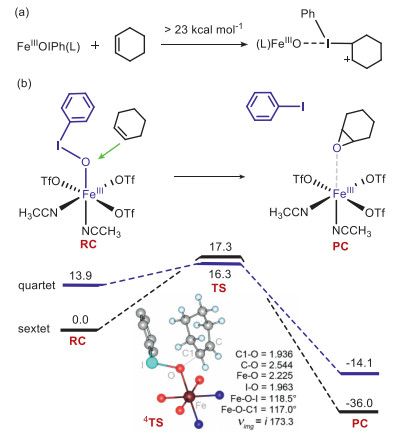
In Valentine's mechanism mentioned above, the carbonium species v is generated from the triangular ⅱ which was obtained by the electrophilic addition of iodine(Ⅲ) to the C=C bond. However, such an electrophilic iodine(Ⅲ) addition was reported to need more than 40 kcal/mol in thioanisole sulfoxidation by a Fe(Ⅲ)-PhIO complex [21]. Therefore, several mechanistic questions arise: Does the triangular iodine(V) species ⅱ exist? How is the carbonium species for the Ritter addition generated? How can one oxidant yield three different products (Besides of the epoxide and the amide products, there was also small amounts of hydroxylated product observed in the experiment) [14]? To answer these questions and continue our mechanistic study of iron-iodosylarene chemistry, cyclohexene oxidation by the iron(Ⅲ)-iodosylarene complex, FeⅢ(PhIO)(OTf)3(CH3CN)2 (1), in Valentine's experiments, was selected as the target of theoretical investigation. Our density functional theory (DFT) calculations at the UB3LYP//B2/B1 level (B1: lanl2dz for Fe, lanl2dzdp for I and 6–31G** for the rest atoms, B2: lanl2tz for Fe, lanl2dzdp for I and 6–31+G** for the rest atoms) demonstrated that 1, instead of the high-valent FeⅣ/Ⅴ=O species 2 or 2′, oxidizes cyclohexene into three products with a good regio-selectivity: C=C bond activation by 1 forms the epoxide product, whereas C−H bond activation generates both the normal hydroxylated product and the unusual amide product, via a novel hydride transfer following by a hydroxyl anion rebound (the hydroxylation route) or a Ritter addition of nitriles (the amide route).
The structure of the employed iron(Ⅲ)-PhIO adduct, FeⅢ(PhIO)(OTf)3(CH3CN)2 (1), is presented in Fig. 1a. The ground state is the sextet state (S = 5/2). The exited quartet/doublet spin states lay higher in energy by 19.3/22.7 kcal/mol (Table S1 in Supporting information). For the ground state 61, the bond distances of Fe–O and I–O are 1.887 Å and 1.927 Å, and the Fe-O-I angle is 126.0°. Then, the conversion of 1 to a high-valent iron−oxo species 2 was investigated. The activation energy is as large as 31.4 kcal/mol. For the low-lying 6TS12, the bond distance of Fe–O and I–O are 1.633 Å and 2.671 Å, respectively, and the Fe−O−I angle is 135.4° The nascent high-valent FeⅣ=O complex 62 lies 32.1 kcal/mol higher than 61. In 2, the spin of the Fe−O moiety is ~3.7 and the PhI moiety is ca. 1.0, thus the O−I bond cleavage is a homolytic process. In short, this conversion is kinetically and thermodynamically unfeasible. Thus, the active oxidant is the Fe(Ⅲ)−iodosylarene species 1, not the high-valent iron-oxo species, which is consistent with Valentine's experimental conclusion [14]. As shown in other metal-iodosylarene systems [18, 21], there is a T-shape halogen bonding interaction between the iodine(Ⅲ) atom of 1 and the approaching substrate. Thus, only one carbon atom can interact with the iodine core and the triangular species ⅱ cannot be formed. The direct electrophilic addition of the C=C bond to form the cyclohexane carbonium complex needs a high activation energy of > 23.0 kcal/mol (Fig. 2a and Fig. S5 in Supporting information). Thus, attribution of the promoted reactivity of the metal-iodosylarene adducts to the hypervalent iodine(Ⅲ) chemistry shown in Scheme 1b is unreasonable. We turned our attention to the traditional electrophilic oxygen addition to the C=C bond via a direct oxygen-atom transfer (DOT) mechanism. The calculated energy profiles are presented in Fig. 2b. Surprisingly, the activation energy is just 16.3 kcal/mol, which is much lower than the one through the electrophilic iodine(Ⅲ) addition. The epoxidation proceeds via a two-state reactivity (TSR) theory [11-13]. A spin inversion occurs from the sextet RC to the quartet TS, followed by a second spin flip from the quartet TS to the sextet PC. For the low-lying 4TS, the Fe−O, I−O and C1−O distances are 2.225 Å, 1.963 Å and 1.936 Å, respectively, and the Fe−O−I and the Fe−O−C1 angles are 118.5° and 117.0°, respectively.

|
Download:
|
| Fig. 1. (a) The geometric information of the FeⅢ(PhIO)(OTf)3(CH3CN)2 species (1). (b) Energy profiles (in kcal/mol) for the conversion of 1 to the high-valent iron-oxo complex 2 species. Key geometric information of the transition states is presented. Hydrogen atoms of PhI are omitted for clarity. Energies are in kcal/mol units, lengths are in Å units, angles are in degree units, and imaginary frequencies are in cm−1 units. | |
{kind=link}

|
Download:
|
| Fig. 2. Energy profiles (in kcal/mol) for along the epoxidation of cyclohexene by 1 species. Hydrogen atoms of PhI are omitted for clarity. Energies are in kcal/mol units, lengths are in Å units, angles are in degree units, and imaginary frequencies are in cm−1 units. | |
{kind=link}
C–H bond activation of cyclohexene by 1 was investigated starting from the same sextet 6RC, and the reaction route of amide formation is presented in Fig. 3. The rate-limiting step is the initial C−H bond activation by 1, holding a barrier of 14.7 kcal/mol. For the low-lying 6TSh, the C2−H distance is 1.192 Å, the O−H one is 1.545 Å, and the C2−H−O angle is 173.7°. Surprisingly, at 6IM1, the H-abstracted substrate (Sub-H) moves far away from the Fe(Ⅲ)−OH moiety (rC2−OH = 4.306 Å, Fig. S8 in Supporting information), suggesting that the Sub-H group escapes from the solvent cage and a non-rebound mechanism [27-29] works. Interestingly, there is no spin for H and the Sub-H moiety at 6IM1, and the Mulliken charge of Sub-H changes from a little negative value (ca. −0.09) at the 6RC state, to a significant positive one (ca. 0.96) at 6IM1 (Fig. S9 and Table S9 in Supporting information). This means that the nascent Sub-H moiety is cationic, which can explain the departure of substrate by the repulsive effect between two cationic parts, i.e., the carbonium and the proton of the Fe−OH moiety. Thus, C–H bond activation of cyclohexene by 1 proceeds via a novel hydride transfer (HT) process [30], which is similar to the C–H bond activation in dehydrogenation of 9, 10-dihydroanthracene by a Mn(Ⅲ)-PhIO complex [18] and Co(Ⅱ)−PhIO2 complex [19]. As Valentine and her coworkers pointed out, a Ritter-like reaction takes place by electrophilic addition of an acetonitrile molecule to the carbonium of Sub-H, with a moderate barrier of 6.1 kcal/mol. After an electron tautomerism, the carbon forms double bond with nitrogen becomes cationic (6IM2). Finally, 6IM2 undergoes a rebound of the hydroxyl anion from the iron core to this carbonium with a barrier of 8.0 kcal/mol, to yield 3-acetamidocyclohexene, which is a key product in Valentine's experiment [14, 16]. We also investigated the hydroxylation route after the initial C−H bond activation started from 6IM1′; that is, the escaped Sub-H moiety can also diffuse back to the solvent cage and locates at the vicinity of the Fe(Ⅲ)–OH complex to form a stable intermediate structure (IMreb) (Fig. S11a in Supporting information). For the ground state 6IMreb (Fig S11b in Supporting information), the distance of C2–OH is 2.435 Å, and the Fe–OH one is 1.823 Å. Subsequently, 6IMreb undergoes a transition state 6TSreb with a tiny barrier of 0.4 kcal/mol, and transfers the hydroxyl to carbonium ion to yield the hydroxylated products. Thus, the hydroxylation is governed by the diffusion rate in a thermodynamics way. Due to a relatively long distance (rC2−OH = 4.916 Å), the entire diffusion process is very slow and the hydroxylation pathway becomes a minor process.

|
Download:
|
| Fig. 3. Energy profiles (in kcal/mol) along the CH bond oxidation of cyclohexene by 1 species. Key geometric information on transition states is presented. Energies are in kcal/mol units, lengths are in Å units, angles are in degree units, and imaginary frequencies are in cm−1 units. | |
{kind=link}
Based on the theoretical findings, we propose a new mechanism for the cyclohexene oxidation by 1 (Scheme 2). When an inorganic salt Fe(OTf)3 is mixed with the iodosylarene polymer, an FeⅢ-PhIO complex (1) is formed, which serves as the active oxidant that oxidizes cyclohexane to afford three different products (the epoxide, the amide and the hydroxylated one) through two reaction pathways: (a) in the electrophilic C=C bond activation, 1 behaves like a normal oxygen donor and shows two-state reactivity to present the epoxide product, via a direct oxygen atom transfer mechanism; (b) in the C−H bond oxidation, the oxidation proceeds via a novel hydride transfer process to form an escaped carbonium species in a non-rebound way, which can continue the reaction via the Ritter mechanism to form an amide by uptake of an acetonitrile to the carbonium moiety, or continue the reaction via a rebound way by a long distance diffusion. The carbonium species returns to the solvent cage with the rebound of the hydroxyl anion to form the hydroxylated product.

|
Download:
|
| Scheme 2. Proposed catalytic mechanism of olefin oxidation involving the FeⅢ−iodosylarene. | |
{kind=link}
In summary, we presented herein the first comprehensive theoretical investigation on the elusive structure-reactivity relationship of the iron(Ⅲ)-iodosylbenzene complex, FeⅢ(PhIO)(OTf)3(CH3CN)2 (1) in Valentine's experiment, which casted the noted one-oxidant mechanism into doubt. The calculated results support Valentine's conclusion that it is the metal-iodosylarene adduct, not the high-valent metal-oxo complex, that serves as the key oxidant. The metal-iodosylarene species 1 was unveiled to behave like a chameleon by adapting its role (a normal oxygen donor, or a two-electron oxidant) when exerts the C=C bond activation to form the epoxide product, or oxidize the C–H bond via a novel hydride transfer mechanism to form an escaped carbonium intermediate, which continues to transform either in a non-rebound way to produce an amide via the Ritter mechanism, or in a rebound way to form a hydroxylated product. Our theoretical work fits and explains Valentine's experimental observations and enrich the non-rebound scenario in the C–H bond activation reactions in bioinorganic chemistry.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (No. 21873052), the Natural Science Foundation of Zhejiang Province (No. LQ20B030004), the Ningbo Natural Science Foundation (No. 202003N4079).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.05.030.
| [1] |
T. Nanjo, E.C. de Lucca, M.C. White, J. Am. Chem. Soc. 139 (2017) 14586-14591. DOI:10.1021/jacs.7b07665 |
| [2] |
H. Sterckx, B. Morel, B.U.W. Maes, Angew. Chem. Int. Ed. 58 (2019) 7946-7970. DOI:10.1002/anie.201804946 |
| [3] |
S. Moncada, R. Gryglewski, S. Bunting, J. Vane, Nature 263 (1976) 663-665. DOI:10.1038/263663a0 |
| [4] |
B.F. Gherman, B.D. Dunietz, D.A. Whittington, S.J. Lippard, R.A. Friesner, J. Am. Chem. Soc. 123 (2001) 3836-3837. DOI:10.1021/ja0055108 |
| [5] |
J. Cho, S. Jeon, S.A. Wilson, et al., Nature 478 (2011) 502-505. DOI:10.1038/nature10535 |
| [6] |
Y.M. Lee, S. Hong, Y. Morimoto, et al., J. Am. Chem. Soc. 132 (2010) 10668-10670. DOI:10.1021/ja103903c |
| [7] |
M.J. Park, J. Lee, Y. Suh, J. Kim, W. Nam, J. Am. Chem. Soc. 128 (2006) 2630-2634. DOI:10.1021/ja055709q |
| [8] |
W. Nam, Account. Chem. Res. 40 (2007) 522-531. DOI:10.1021/ar700027f |
| [9] |
D. Kass, T. Corona, K. Warm, et al., J. Am. Chem. Soc. 142 (2020) 5924-5928. DOI:10.1021/jacs.9b13756 |
| [10] |
J.T. Groves, G.A. McClusky, Biochem. Biophys. Res. Commun. 81 (1978) 154-160. DOI:10.1016/0006-291X(78)91643-1 |
| [11] |
S. Shaik, M. Filatov, D. Schröder, H. Schwarz, Chem. Eur. J. 4 (1998) 193-199. DOI:10.1002/(SICI)1521-3765(19980210)4:2<193::AID-CHEM193>3.0.CO;2-Q |
| [12] |
F. Ogliaro, N. Harris, S. Cohen, et al., J. Am. Chem. Soc. 122 (2000) 8977-8989. DOI:10.1021/ja991878x |
| [13] |
S. Shaik, S.P. de Visser, F. Ogliaro, H. Schwarz, D. Schröder, Curr. Opin. Chem. Biol. 6 (2002) 556-567. DOI:10.1016/S1367-5931(02)00363-0 |
| [14] |
Y. Yang, F. Diederich, J.S. Valentine, J. Am. Chem. Soc. 112 (1990) 7826-7828. DOI:10.1021/ja00177a071 |
| [15] |
W. Nam, J.S. Valentine, J. Am. Chem. Soc. 112 (1990) 4977-4979. DOI:10.1021/ja00168a063 |
| [16] |
Y. Yang, F. Diederich, J.S. Valentine, J. Am. Chem. Soc. 113 (1991) 7195-7205. DOI:10.1021/ja00019a016 |
| [17] |
W. Nam, S.W. Jin, M.H. Lim, J.Y. Ryu, C. Kim, Inorg. Chem. 41 (2002) 3647-3652. DOI:10.1021/ic011145p |
| [18] |
D. Sun, X. Chen, L. Gao, Y. Zhao, Y. Wang, Front. Chem. 8 (2020) 744. DOI:10.3389/fchem.2020.00744 |
| [19] |
X. Chen, D. Sun, L. Gao, et al., Chem. Commun. 57 (2021) 3115-3118. DOI:10.1039/d0cc07894h |
| [20] |
M. Guo, Y.M. Lee, M.S. Seo, et al., Inorg. Chem. 57 (2018) 10232-10240. DOI:10.1021/acs.inorgchem.8b01426 |
| [21] |
Y. Kang, X.X. Li, K.B. Cho, et al., J. Am. Chem. Soc. 139 (2017) 7444-7447. DOI:10.1021/jacs.7b03310 |
| [22] |
J. Yang, M.S. Seo, K.H. Kim, et al., Angew. Chem. Int. Ed. 59 (2020) 13581-13585. DOI:10.1002/anie.202005091 |
| [23] |
E.A. Hill, M.L. Kelty, A.S. Filatov, J.S. Anderson, Chem. Sci. 9 (2018) 4493-4499. DOI:10.1039/c8sc01167b |
| [24] |
M. Guo, Y.M. Lee, S. Fukuzumi, W. Nam, Coord. Chem. Rev. 435 (2021) 213807. DOI:10.1016/j.ccr.2021.213807 |
| [25] |
J.J. Ritter, P.P. Minieri, J. Am. Chem. Soc. 70 (1948) 4045-4048. DOI:10.1021/ja01192a022 |
| [26] |
J.J. Ritter, J. Kalish, J. Am. Chem. Soc. 70 (1948) 4048-4050. DOI:10.1021/ja01192a023 |
| [27] |
E. Kwon, K.B. Cho, S. Hong, W. Nam, Chem. Commun. 50 (2014) 5572-5575. DOI:10.1039/C4CC01058B |
| [28] |
S. Fukuzumi, K.B. Cho, Y.M. Lee, S. Hong, W. Nam, Chem. Soc. Rev. 49 (2020) 8988-9027. DOI:10.1039/d0cs01251c |
| [29] |
K.B. Cho, H. Hirao, S. Shaik, W. Nam, Chem. Soc. Rev. 45 (2016) 1197-1210. DOI:10.1039/C5CS00566C |
| [30] |
J. Li, S. Zhou, M. Schlangen, T. Weiske, H. Schwarz, Angew. Chem. Int. Ed. 55 (2016) 13072-13075. DOI:10.1002/anie.201606707 |