 2021, Vol. 32
2021, Vol. 32 


b Renewable and Sustainable Energy Institute, and Department of Chemical and Biological Engineering, University of Colorado Boulder, Boulder, CO 80309, United States
Covalent organic frameworks (COFs) represent a class of porous and crystalline polymers that have been rapidly developed and widely used for various applications, such as gas storage and separation, heterogeneous catalysis, sensing, and energy storage [1-7]. Reversible linkages such as boronic ester and imine bonds, with self-correction mechanism enabled, have been extensively used for COF synthesis. Because of the vulnerable nature of the reversible bonds, these materials usually show considerable sensitivity to moisture and acidic/basic conditions. Although some of them could exhibit enhanced chemical stability by using bulky protective groups, tautomerization or intramolecular hydrogen bonds [8-13], their applications under harsh conditions, particularly under strong acidic or basic conditions, still represent a challenge. In recent years, some robust irreversible linkages, such as dioxin and phenazine, have been utilized to construct COFs, which enable high stability of the frameworks even under harsh conditions [14-19], offering the potential applications of these robust COFs in chemical or electrochemical catalysis at high temperature and/or in strong acidic or basic medium.
In this work, we synthesized a series of dioxin-linked metallated phthalocyanine based COFs (MPc-dx-COFs) and investigated their potential applications as electrocatalysts for the oxygen reduction reaction (ORR). The two-dimensional planar frameworks consisting of metallated phthalocyanine active sites and 1, 4-dioxin linkages have an average pore size of 2.0 nm. Connected by robust irreversible dioxin bonds, the COF materials are stable under various harsh conditions including boiling water, concentrated acid (12 mol/L HCl), base (12 mol/L NaOH), oxidative (30% H2O2) and reductive (1 mol/L NaBH4) solutions. The as-synthesized frameworks also exhibit catalytic activity for ORR. They can selectively reduce oxygen into hydroxyl ions through a four-electron pathway upon mixing with Vulcan carbon to assist with conductivity. To the best of our knowledge, this presents the first example of direct use of metallophthalocyanine COFs in ORR electrocatalysis.
The CoPc-dx-COF was synthesized via nucleophilic aromatic substitution (SNAr) between the Co(II)-metallated octahydroxyphthalocyanine and tetrafluoroterephthalonitrile (TFPN) in 1, 4-dioxane at 120 ℃ for 3 days. Triethylamine (NEt3) was used as the base, which deprotonates the hydroxy groups and neutralizes the HF byproduct generated from the reaction (Scheme 1). The product was collected and washed with deionized water, methanol, dichloromethane, and acetone. The product was further purified by sequentially washing in Soxhlet extractor with methanol and acetone. The CoPc-dx-COF was dried in a vacuum oven at 120 ℃ overnight to give a dark green solid with an 81% yield. Likewise, ZnPc-dx-COF and H2Pc-dx-COF (non-metallated) were synthesized using the same method but different octahydroxyphthalocyanine as the starting materials. It should be noted that the dioxin linkages cannot be cleaved and reformed under the applied reaction condition for the COF synthesis. Even after stirring at 120 ℃ for 24 h, no exchange reaction was observed, indicating the irreversible robust nature of the dioxin linkage (Scheme 1b, Fig. S1 in Supporting information). This result is also consistent with the previous literature report [14].

|
Download:
|
| Scheme 1. Synthesis of CoPc-dx-COF via (a) nucleophilic reaction and (b) irreversible dioxin exchange. | |
{kind=link}
Successful synthesis of the frameworks through SNAr reaction forming 1, 4-dioxine linkages was confirmed using various characterization techniques. The 13C cross-polarization magic angle spinning (CP-MAS) NMR spectrum of the H2Pc-dx-COF shows the peaks around 110 ppm and ~145 ppm corresponding to the nitrile carbons and dioxin carbons, respectively (Fig. S2 in Supporting information). Other aromatic protons show the resonance signals at around 130 ppm. Fourier-Transform Infrared spectroscopy (FT-IR) spectra (Fig. S3 in Supporting information) of the purified MPc-dx-COFs showed the characteristic dioxin C-O asymmetric and symmetric stretching bands around 1270 and 1000 cm-1, which indicates the formation of 1, 4-dioxin bonds. The characteristic band around 2230 cm-1 indicates the presence of cyano groups. The disappearance of O-H stretching band between 3200 cm-1 and 3400 cm-1 indicates the full conversion of hydroxy groups in the phthalocyanine starting material. Scanning electron microscope (SEM) images revealed the morphologies of the MPc-dx-COFs, all showing anomalous particles of different sizes.
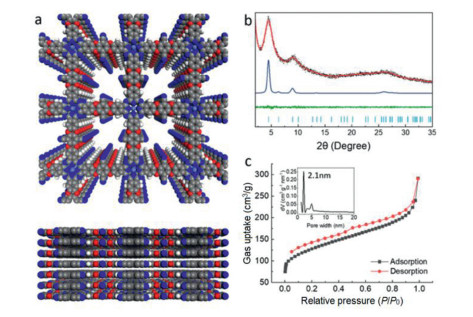
The crystallinities of CoPc-dx-COFs were characterized by powder X-ray diffraction (PXRD) analysis. Fig. 1b shows an intensive peak at 2θ = 4.48° corresponding to (100) reflection planes, two small peaks at 2θ = 6.34° and 8.97°, and a broad peak at 2θ = 25.49° correspond to (110), (200) and (001) reflection planes, respectively. We did not observe any diffraction peaks corresponding to the starting materials (Fig. S4 in Supporting information). The structure simulation (Fig. 1a) and Pawley refinement of the PXRD pattern profile were performed using Material Studio. A simulated AA eclipsed model was built as a 2D layered structure in a tetragonal system. After energy minimization, the unit cell parameters were given as a = b = 20.33 Å; c = 3.44 Å. Similarly, the AB staggered model was built, which gives the same parameters of a and b but c = 6.88 Å (Fig. S5b in Supporting information). The experimental data match well with the simulated PXRD patterns of eclipsed AA stacking of the COF layers. Pawley refinement gives Rwp = 5.08% and Rp = 3.82% with the eclipsed structure. Therefore, we determined that the COFs adopt the fully eclipsed AA stacking mode. As shown in Figs. S6a and b (Supporting information), ZnPc-dx-COF and H2Pc-dx-COF have similar lattice parameters as CoPc-dx-COF. We observed a type II nitrogen adsorption isotherm for CoPc-dx-COF. Brunauer-Emmett-Teller (BET) surface area of CoPc-dx-COF was calculated to be 417 m2/g. The pore size distribution of the framework was estimated using nonlocal density functional theory (NL-DFT), which shows mesopores of the size of 21 Å (Fig. 1c), consistent with the simulated AA consistent with the simulated AA stacking model. ZnPc-dx-COF and H2Pc-dx-COF also gave type II nitrogen adsorption isotherms but with smaller surface area (Figs. S6c and d in Supporting information).

|
Download:
|
| Fig. 1. Characterizations of the CoPc-dx-COF: (a) Top view and side view of CoPc-dx-COF in simulated AA stacking mode (light blue, M; grey, C; blue, N; white, H; red, O). (b) Experimental PXRD profile of CoPc-dx-COF (black), Pawley refined profile (red), their difference (green), simulation results based on AA stacking (blue) and its reflection (yellow). (c) Nitrogen sorption isotherm profile at 77 K and pore size distribution of CoPc-dx-COF. | |
{kind=link}
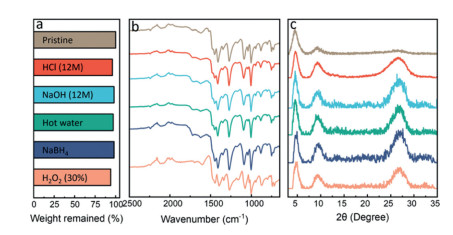
We next demonstrated the exceptionally high stability of CoPc-dx-COF under various harsh conditions. The chemical stability of CoPc-dx-COF was studied by soaking the material in various solutions, including 30% H2O2, 1 mol/L NaBH4, hot water (90 ℃), concentrated HCl (12 mol/L), and NaOH (12 mol/L) for three days. As shown in Fig. 2a, CoPc-dx-COFs only experienced 2 wt%-6 wt% weight loss under such extreme conditions. Only 4 wt% of weight loss occurred in 12 mol/L HCl. In 12 mol/L NaOH and hot water, the weight losses were 3% and 2% respectively. Under reductive and oxidative conditions, the weight losses were 2% and 6%, respectively. As shown in the FTIR spectra (Fig. 2b), the absorption signals of the samples are almost identical, only showing minor differences, which indicates there is negligible damage in their chemical structures after the treatments. We attribute the excellent stability of CoPc-dx-COF to the robustness of the 1, 4-dioxin linkages and phthalocyanine rings. The PXRD diffraction patterns changed noticeably after the treatments as shown in Fig. 2c. Although the width at half height of the first two peaks does not show any obvious change compared to the original sample, the intensity of the peak around 26° increased quite a bit, which might be caused by the re-stacking of COFs layers.

|
Download:
|
| Fig. 2. Stability test of CoPc-dx-COF: (a) wt% of the COFs remained after different treatments; (b) FT-IR spectra and (c) PXRD profilesof the COF samples after different treatments. | |
{kind=link}
To date, although there has been significant progress in the study of metallated phthalocyanine and porphyrin based electrocatalysts [20-24], applications of as-synthesized COFs and covalently bonded polymers with well-defined active site coordination (for example, in catalysis) still remain limited due to the synthetic challenges in creating these materials to simultaneously possess both ordered structures and stabilities [25-30]. Here we have considered, as a first step toward this goal, linking the active catalytic sites with robust bonds in order to enhance the stability of the molecular catalyst units. Doing so in a regular, porous COF structures may in principle further permit access to all of the metal centers, provided that the overall diffusion paths are kept short so that internal mass transfer does not become a limitation. Given the highly stable crystalline MPc-dx-COFs, we next evaluated these metallated phthalocyanine COFs as electrocatalysts for oxygen reduction reaction (ORR).
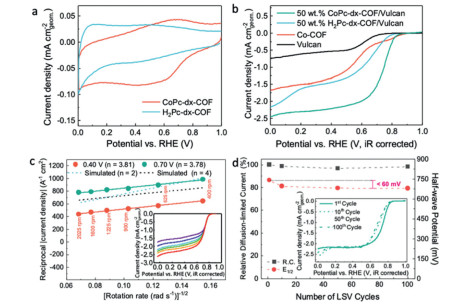
First, the electrochemical behavior of these catalysts in the absence of O2 was evaluated by cyclic voltammetry (CV) in argon-saturated 0.1 mol/L KOH (Fig. 3a). We assign the electrochemical waves around 0.65 V to the redox activities of the metallic centers (Co3+/Co2+) since they are absent in the CV curve of the H2Pc-dx-COF. Initial tests at higher currents revealed conductivity limitations, so we next mixed CoPc-dx-COF materials with Vulcan carbon in different proportions to increase the conductivity for investigation of the catalytic activity toward ORR under various conditions. For clarity, we name these samples such that, for example, 50 wt% COF of COF/Vulcan mixture with a loading of 50.0 µg/cm2 indicates the catalyst layer contains 25.0 µg of the COF per cm2. Linear sweep voltammetry (LSV) was performed using a rotating disk electrode (RDE) in O2-saturated 0.1 mol/L KOH aqueous solution for five samples with different mass loadings of the COF alone (12.5 and 50.0 µg/cm2) and 50 wt% COF/Vulcan mixtures (12.5, 25.0 and 50.0 µg/cm2, i.e., catalyst layers with 6.25, 12.5 and 25.0 µg/cm2 of COF, Fig. S7a in Supporting information). The diffusion-limited current densities at 0.2 V vs. RHE for 12.5 and 50.0 µg/cm2 COF were much lower than that observed for 12.5 µg/cm2 loading of 50 wt% COF/Vulcan (containing 6.25 µg/cm2 of the COF), indicating the conductivity is a crucial parameter for the activity of these catalysts. It should be noted the Vulcan carbon alone shows a very low catalytic activity (much lower than even the pure COF at the same mass loading). Thus, the activity enhancement for the 50 wt% COF/Vulcan is not attributable to the electrochemical activity of Vulcan; it is due to increased conductivity to active sites on the COF. In the absence of Vulcan, the sample with the lower loading of the COF (12.5 µg/cm2) shows a slightly higher activity compared to the higher loading of pure COF (50.0 µg/cm2), further illustrating that resistance is a limiting factor for the pure COF.

|
Download:
|
| Fig. 3. Electrochemical tests of CoPc-dx-COF: (a) Cyclic voltammograms of H2Pc-dx-COF and CoPc-dx-COF at 0.05 mg/cm2 loading and a scan rate of 100 mV/s in Ar-saturated 0.1 mol/L KOH. (b) Linear sweep voltammograms (LSV) in O2-saturated 0.1 mol/L KOH at a scan rate of 10 mV/s for different materials loading. (c) Koutecky-Levich plots for 50 wt% CoPc-dx-COF/Vulcan catalyst (25 µg/cm2) at 0.7 and 0.4 V vs. RHE. Inset: LSV in O2-saturated 0.1 mol/L KOH at a scan rate of 10 mV/s for different rotation rates. (d) Stability tests for 50 wt% CoPc-dx-COF/Vulcan, loading 25 µg/cm2. R.C. stands for diffusion-limited current density. | |
{kind=link}
Optimization of the COF/Vulcan ratio is illustrated in Fig. S7 (Supporting information), where it was found that a 1:1 ratio best balances the conductivity against dilution of active sites. To determine an appropriate film thickness (where diffusion into the film is not limiting), we also varied the absolute loading (Fig. S7) from 12.5 µg/cm2 to 50.0 µg/cm2 and found diminishing returns beyond 25.0 µg/cm2, indicating this loading gave sufficient density of active sites on the electrode surface while not creating diffusionally-inaccessible regions. As shown in Fig. 3b, under the optimized conditions, CoPc-dx-COF/Vulcan mixture shows the best reactivity among Vulcan, CoPc-dx-COF, and H2Pc-COF/Vulcan catalysts. The result confirms that the coordinated metallic centers are the most active sites for ORR in these materials. In Fig. 3c, the K-L plots at 0.7 V (the region controlled by diffusion and kinetic phenomena) and 0.4 V (the region at diffusion-controlled regime) manifested good linearity, and the number of electrons involved in the reduction process was estimated as ~3.8 from Eq. S1, showing that the ORR mainly follows the four-electron path, i.e., O2 is reduced to OH-. This result was compared with rotating ring-disk electrode (RRDE) experiments performed on 50 wt% CoPc-dx-COF/Vulcan, 50 wt% ZnPc-dx-COF/Vulcan, and Vulcan inks deposited on the disc for extra evaluation of the ORR selectivity. Before ORR-RRDE experiments, RRDE collection efficiency was determined, obtaining a 20% efficiency, as is shown in Fig. S9 (Supporting information). As can be seen in Fig. S10 (Supporting information), the 2e- path―O2 reduction to H2O2 (or HO2- ions)―contributed with only 10% of the total current for COFs, and the average number of electrons for these catalysts was 3.8, corroborating with K-L results. This 2e- path background current density may be associated with the exposed Vulcan sites, which showed a higher H2O2 production (30% of the total current) than the COFs (Fig. S9).
Electrochemical stability experiments (Fig. 3d and Fig. S11 in Supporting information) showed that 50 wt% CoPc-dx-COF/Vulcan has good stability; it was observed less than 60 mV shift in half-wave potential (E1/2) after 100 cycles, and < 3% decrease in diffusion-limited current density, indicating retention of selectivity to the four-electron path. In addition, the activity losses are mainly associated with break-in, and the polarization curve becomes very stable after the 10th cycle; it remains much higher than observed for Vulcan and H2Pc-COF/Vulcan catalysts.
In conclusion, we have successfully synthesized the COF materials containing metallated phthalocyanine catalytic sites connected with robust 1, 4-dioxin linkages by using nucleophilic aromatic substitution reaction. These COFs show remarkable chemical stability even under harsh conditions, e.g., soaking in strong acid (12 mol/L aq. HCl), base (12 mol/L aq. NaOH), oxidizing agent (30% H2O2), reducing agent (1 mol/L NaBH4), or hot water (90 ℃) for three days. These COFs can be directly used as electrocatalysts for the oxygen reduction reaction upon mixing with Vulcan without being converted to metal- and nitrogen-carbon-based (M-N-C) materials through pyrolysis as commonly used in previous studies. We envision that stable COFs consisting of catalytically active building blocks and robust linkages could open new possibilities for developing highly efficient catalysts targeting important environment and energy applications.
Declaration of competing interestThere is no conflict to declare.
AcknowledgmentsThis work was supported by University of Colorado Boulder and K. C. Wong Education Foundation. Z. Lei acknowledges financial support by the Koch Graduate fellowship.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.04.047.
| [1] |
A.P. Cote, A.I. Benin, N.W. Ockwig, et al., Science 310 (2005) 1166-1170. DOI:10.1126/science.1120411 |
| [2] |
X. Feng, X. Ding, D. Jiang, Chem. Soc. Rev. 41 (2012) 6010-6022. DOI:10.1039/c2cs35157a |
| [3] |
S.Y. Ding, W. Wang, Chem. Soc. Rev. 42 (2013) 548-568. DOI:10.1039/C2CS35072F |
| [4] |
E. Jin, M. Asada, Q. Xu, et al., Science (2017) 673-676. |
| [5] |
K. Geng, T. He, R. Liu, et al., Chem. Rev. 120 (2020) 8814-8933. DOI:10.1021/acs.chemrev.9b00550 |
| [6] |
Z. Wang, S. Zhang, Y. Chen, Z. Zhang, S. Ma, Chem. Soc. Rev. 49 (2020) 708-735. DOI:10.1039/C9CS00827F |
| [7] |
X. Guan, F. Chen, Q. Fang, S. Qiu, Chem. Soc. Rev. 49 (2020) 1357-1384. DOI:10.1039/C9CS00911F |
| [8] |
L.M. Lanni, R.W. Tilford, M. Bharathy, J.J. Lavigne, J. Am. Chem. Soc. 133 (2011) 13975-13983. DOI:10.1021/ja203807h |
| [9] |
S. Kandambeth, A. Mallick, B. Lukose, et al., J. Am. Chem. Soc. 134 (2012) 19524-19527. DOI:10.1021/ja308278w |
| [10] |
S. Kandambeth, D.B. Shinde, M.K. Panda, et al., Angew. Chem. Int. Ed. Engl. 52 (2013) 13052-13056. DOI:10.1002/anie.201306775 |
| [11] |
H. Xu, J. Gao, D. Jiang, Nat. Chem. 7 (2015) 905-912. DOI:10.1038/nchem.2352 |
| [12] |
P.F. Wei, M.Z. Qi, Z.P. Wang, et al., J. Am. Chem. Soc. 140 (2018) 4623-4631. DOI:10.1021/jacs.8b00571 |
| [13] |
Y. Ma, Y. Wang, H. Li, et al., Angew. Chem. Int. Ed. Engl. 59 (2020) 19633-19638. DOI:10.1002/anie.202005277 |
| [14] |
B. Zhang, M. Wei, H. Mao, et al., J. Am. Chem. Soc. 140 (2018) 12715-12719. DOI:10.1021/jacs.8b08374 |
| [15] |
Z. Meng, R.M. Stolz, K.A. Mirica, J. Am. Chem. Soc. 141 (2019) 11929-11937. DOI:10.1021/jacs.9b03441 |
| [16] |
X. Guan, H. Li, Y. Ma, et al., Nat. Chem. 11 (2019) 587-594. DOI:10.1038/s41557-019-0238-5 |
| [17] |
M. Wang, M. Ballabio, M. Wang, et al., J. Am. Chem. Soc. 141 (2019) 16810-16816. DOI:10.1021/jacs.9b07644 |
| [18] |
N. Huang, K.H. Lee, Y. Yue, et al., Angew. Chem. Int. Ed. Engl. (2020) 16587-16593. |
| [19] |
M. Lu, M. Zhang, C.G. Liu, et al., Angew. Chem. Int. Ed. Engl. 60 (2021) 4864-4871. DOI:10.1002/anie.202011722 |
| [20] |
B. Lv, X. Li, K. Guo, et al., Angew. Chem. Int. Ed. Engl. 60 (2021) 12742-12746. DOI:10.1002/anie.202102523 |
| [21] |
H. Lv, H. Guo, K. Guo, et al., Chin. Chem. Lett. 32 (2021) 2841-2845. DOI:10.1016/j.cclet.2021.02.032 |
| [22] |
D. Kim, M. Kang, H. Ha, C.S. Hong, M. Kim, Coord. Chem. Rev. 438 (2021) 213892. DOI:10.1016/j.ccr.2021.213892 |
| [23] |
Z. Liang, H. Guo, G. Zhou, et al., Angew. Chem. Int. Ed. 60 (2021) 8472-8476. DOI:10.1002/anie.202016024 |
| [24] |
L. Xie, X.P. Zhang, B. Zhao, et al., Angew. Chem. Int. Ed. (2021) 7654-7659. |
| [25] |
G. Lu, H. Yang, Y. Zhu, et al., J. Mater. Chem. A 3 (2015) 4954-4959. DOI:10.1039/C4TA06231K |
| [26] |
S. Lu, Y. Jin, H. Gu, W. Zhang, Sci. China Chem. (2017) 999-1006. |
| [27] |
W. Liu, K. Wang, C. Wang, et al., J. Mater. Chem. A 6 (2018) 22851-22857. DOI:10.1039/C8TA08173E |
| [28] |
R. Chen, J.L. Shi, Y. Ma, et al., Angew. Chem. Int. Ed. 58 (2019) 6430-6434. DOI:10.1002/anie.201902543 |
| [29] |
D. Li, C. Li, L. Zhang, et al., J. Am. Chem. Soc. 142 (2020) 8104-8108. DOI:10.1021/jacs.0c02225 |
| [30] |
R. Chen, Y. Wang, Y. Ma, et al., Nat. Commun. 12 (2021) 1354. DOI:10.1038/s41467-021-21527-3 |