 2021, Vol. 32
2021, Vol. 32 


b School of Materials and Chemical Engineering, Ningbo University of Technology, Ningbo 315211, China
The charge transport process in proteins across peptide segments has been proved to be efficient over long distance in both solution state [1-3] and solid state [4-8] via charge tunneling through an optimistic pathway, initiating the hope on bio-electronics using protein and peptide as functional units [9-14]. The charge tunneling over homopeptides with different side groups shows variation in junction current which indicates the tunability of charge transport across peptide junctions by choice of different amino acids side groups [15-17]. Experimental studies and theoretical calculations on hetero-peptides, on the other hand, prove a series tunneling charge transport process and provide a new strategy to modify charge transport by conjugated side group doping in homopeptides, which tunes the tunneling barrier landscape in the junction conductance, leading to a facile modulation in the junction conductance [18, 19].
Histidine, among all amino acids with conjugate side groups, is more frequently found in oxidoreductase enzymes than amino acids with non-conjugating side groups [20]. The imidazole side group of histidine can be protonated/deprotonated [21, 22] and facilitates proton coupled electron transport in photosynthesis systems [23]. Histidine has been proposed to serve as the last site of the tunneling pathway via electron tunnels through proteins to metal ions of the redox center [24]. Recent study on homopeptides of histidine found similar tunneling attenuation factor comparable to conjugated molecules (~0.5 Å-1), indicating histidine is an efficient tunneling mediator. The properties of histidine suggest a promising candidate for modification of peptide through multiple aspects [25]. While the tunability of peptide energy barrier landscape and junction tunneling by histidine doping remains an unknown question.
In this work, we prepared peptide self-assembled monolayers of similar thickness with histidine doping into oligo-alanine peptides at different position. EGaIn was used for the study of the charge transport across these peptide monolayers (see schematic experimental setup in Fig. 1). The doping of histidine into 7-alanine peptides increased the junction tunneling current, indicating effective modification of the peptide junction energy landscape. Based on theoretical modeling combined with energy state analysis by both density-functional theory (DFT) calculation and ultraviolet photoelectron spectroscopy (UPS), we found series tunneling through tested peptide junctions and doping histidine effectively reduced the junction tunneling barrier height. The sequence of histidine doped into 7-alanine peptides resulted in different energy landscape, thus achieving fine tuning of the charge transport through peptide monolayers.

|
Download:
|
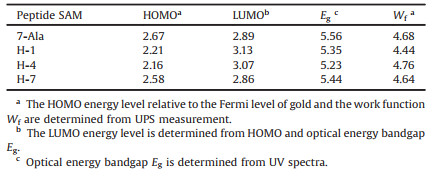
| Fig. 1. Molecular structures of peptides in this work. 7-Ala: MPA-AAAAAAA, H-1: MPA-HAAAAAA, H-4: MPA-AAAHAAA, H-7: MPA-AAAAAAH. (A = alanine, H = histidine) Schematic illustration of the experimental setup of the EGaIn I-V measurement. | |
In order to study the effect of histidine doping in peptides, we designed a series of peptide derivatives including hepta-alanine (7-Ala) and histidine substitution at the N-terminal (H-1), in the center (H-4) and at the C-terminal (H-7) of the hepta-alanine (see chemical structure in Fig. 1) with a mercaptopropionic acid (MPA) connected to the peptides through covalent bond at the N terminus for self-assembling to the gold substrate by S-Au bond.
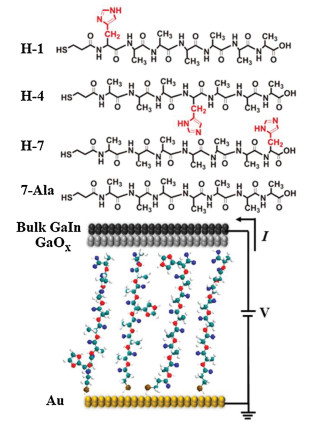
These four types of peptides (7-Ala, H-1, H-4 and H-7) were all immobilized onto the gold substrate through self-assembling process in solution and the self-assembled monolayers (SAMs) of similar thickness of ~2 nm for these peptides were obtained by ellipsometry measurement (Fig. 2a). The theoretical length of these designed peptide derivatives with fully stretched conformation is ~3 nm, while measured SAMs thickness is around 2 nm, indicating the peptide chains may take a random coiled conformation as commonly seen in short oligopeptides, such as histidine or tryptophan peptides, on the gold substrate [18, 25]. In order to gain further information on the peptide SAM structure, we conducted atomic force scratching, cyclic voltametry desorption and Infrared spectroscopy (IR) measurment. Atomic force microscopy (AFM) characterization of the mophology of the SAMs found an uniform peptide monolayer without aggregation and the thickness of all peptide SAMs measured by AFM scratch agrees well with ellipsometry thickness (Fig. 2a, AFM images of peptide SAMs and bare gold substrate can be seen in Supporting information). We further conducted electrochemical desorption experiment for all four types of peptide SAMs to calculate the surface coverage and similar values (~ 1.0 × 10-10 mol/cm2) for H-1, H-4 and 7-Ala SAMs and slightly larger surface coverage for H-7 SAM (~1.3 × 10-10 mol/cm2) were found. The larger surface coverage of H-7 and the single desorption peak in CV occuring at more negative potential (Fig. S3 in Supporting information) indicates more densely packed molecules in the SAM, thus resulting a larger SAM thickness for H-7 compared to the other peptide SAMs [26].

|
Download:
|
| Fig. 2. (a) The ellipsometry and AFM scratch measured thickness of 7-Ala, H-1, H-4 and H-7 SAMs and the surface coverage of the corresponding SAMs. (b) Normalized reflective IR of 7-Ala, H-1, H-4 and H-7 SAMs immobilized on gold substrate. | |
In order to study the peptide orientation in the monolayers, we conducted reflective IR measurement on the peptide SAMs immobilized on the gold surface. The IR spectra of 7-Ala, H-1, H-4 and H-7 all show two amide peaks (amide Ⅰ at ~1668 cm-1 and amide Ⅱ at ~1542 cm-1) which confirms the presence of the peptides on the gold substrate (Fig. 2b). The conformation and amide chain orientation can be interpreted from the reflective IR, where only vibration modes with a transition dipole perpendicular to the meatal surface are active in the reflective IR. Transition dipole moments of amide Ⅰ and amide Ⅱ in the amide plane are nearly along and perpendicular to the peptide chain, respectively. The orientation of the amide plane relative to the gold surface thus can be deduced from these two amide dipole orientation by the relative intensity of two amide peaks from IR absorbance [27]. When the relative intensity of amide Ⅰ to amide Ⅱ is larger, the SAM on the gold surface is less tilted [28]. H-1, H-7 and 7-Ala SAMs all exhibit a large amide Ⅰ/amide Ⅱ peak intensity ratio compared to that of H-4 SAM, which indicates H-1, H-7 and 7-Ala have smaller tilting angle on the gold substrate, which might originate from a more stretched conformation, while H-4 SAM has a larger tilting angle due to more collapsed conformation on the gold substrate. These results are consistent with larger thickness for H-1, H-7 and 7-Ala monolayers and smaller thickness for H-4.
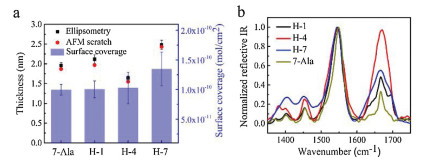
We then turn to study the charge tranpsort properties of the monolayer by applying I–V measurement. We used gold substrate of the monolayers as bottom electrode and EGaIn as top electrode. The EGaIn is known as soft and non-permeable electrode for SAMs based molecular junction characterization due to its liquid nature and naturally produced GaO protective layer [29-31]. The measurement was performed by a home-made EGaIn system at room temperature and low humidity in the dry air. The 2D histograms of log(J)-V curves for 7-Ala, H-1, H-4 and H-7 molecular junctions are illustrated in Figs. 3a-d, where J is the current density. All peptide junctions showed linear I–V relation in the low bias region and non-linear I–V curve in the high bias region, which is believed to be tunneling charge transport (see Fig. 4 for averaged I–V curves of 7-Ala, H-1, H-4 and H-7 molecular junctions). Histidine doping into oligo-alanine peptide exhibits obvious tuning on the current density of the peptide junctions as big as one order of magnitude, following a variation in current density as: H-4 > H-1 > H-7 ~ 7-Ala (Fig. 3e).

|
Download:
|
| Fig. 3. (a-d) The 2D histogram of log(J) distribution of 7-Ala, H-1, H-4 and H-7 junctions. (e) Average log(J)-V curves of the tested peptide junctions of 7-Ala, H-1, H-4 and H-7 SAMs. (f) Junction charge tunneling distance vs. log(J) at 0.05 V of the peptide junctions of 7-Ala, H-1, H-4 and H-7 SAMs. | |

|
Download:
|
| Fig. 4. Taylor expansion fitted I-V curves and experimental I-V curves of peptide junctions of (a) 7-Ala, (b) H-1, (c) H-4 and (d) H-7. | |
For the low bias range, it has been reported that tunneling current across the polypeptide can be expressed as charge transport over heterogeneous energy barrier with series of sub-units in a "series tunneling" manner, as shown in Eq. 1 below [32]:
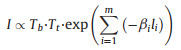
|
(1) |
where βi and li are the transport attenuation factor and length of individual subunits, respectively. Tb and Tt describe the tunneling probability across the bottom and top electrode-peptide interface, respectively. β is characteristic parameter that describes how fast the conductance decay with length. In the simple Simmons model, it relates to the tunneling barrier height εh as Eq. 2 [15]:
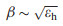
|
(2) |
Thus a smaller barrier will produce a smaller decay coefficient and afford a more efficient long range tunneling. We extracted the low bias conductance of the 4 peptides, and summarized them with respective to film thickness in Fig. 3f. We can see an obvious increase in the current density magnitude of H-1 compared to 7-Ala molecular junction at similar thickness. According to Eqs. 1 and 2, this implies a smaller β and lower tunnelign barrier εh. For H-7, the monolayer thickness is significatly larger than 7-Ala, while the conductance is similar, again implying better long range tunneling by histidine doping. Since the thickness of H-4 is smaller compared to 7-Ala, we cannot separate the influence of conductance increase from the two factors of smaller thickness and the change in tunneling barrier height. In our previous study, based on homo-histidine peptide junctions, we obtained a distance attenuation factor of the histidine of β ~ 0.5 Å-1, which is similar to the value obtained from unsaturated molecules, indicating the effective charge tunneling across oligo-histidine junctions [25]. Thus we propose that the doping of hisitidine unit into oligo-alanine peptide reduces the tunneling energy barrier.
To further understand the histidine doping effect we did more detailed quantitative anaylsis of junction I-V characteristics using the transition voltage spectrum (TVS), which has been widely used as quantification tool for molecular transport characteristics, due to its simplicity and reproducibility. The tunneling barrier height (εh) can be correlated to transition voltage (VTVS) by Eq. 3 [33]:

|
(3) |
However, the limitation of this method is that one has to scan the bias to high voltage to obtain the VTVS. In this work, we cannot apply high bias to the peptide SAMs due to its stability. Therefore we turn to a method using I-V simulation parameters to get the VTVS. Ayelet Vilan and coworkers ever proposed a method to derive VTVS using a scaling model based on Taylor expansion of the current in response to voltage. According to Vilan, the I-V curves in tunneling junction have approximately a generic parabolic shape, which can be expended using Taylor expansion as shown below.
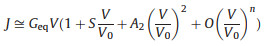
|
(4) |
where Geq is the equilibrium conductance (i.e., the conductance at V →0), S measures the even function component of the I-V in the transport and can generally be omitted. A2 is a pre-factor describing the symmetry of junction current which equals 1 for symmetric junctions and weakly varies with asymmetry. O represents higher order terms in Taylor expansion for current density. The key parameter in Eq. 4 is V0, which is the bias scaling parameter. Vilan showed that the transition voltage VTVS is related to V0 by Eq. 5 [34]:
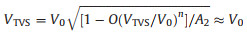
|
(5) |
Therefore, by fitting I-V curves using Eq. 4, one can obtain V0, and then find out the transition voltage VTVS and tunneling energy barrier εh. This method largely expands the use of TVS in cases where the transition voltage has not been reached. We utilized this method here to study the I-V data of the peptide junctions and obtained well agreed fitting curves (Fig. 4 for the fitting results and Supporting information for details).
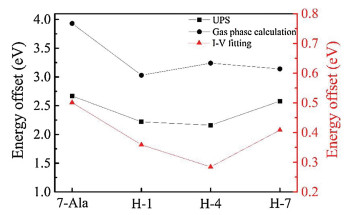
From the fitting obtained value of V0 we could find VTVS derived junction energy barrier εh. The doping of histidine into the oligo-alanine lowered the tunneling energy barrier in the peptide junctions. (Fig. 5, the energy barrier of the peptide junctions is summarized in Table 1). Moreover, we also find the energy barrier change varies as the position of the histidine in the oligo-alanine chain. Thus, we propose that histidine doping effect on junction energy barrier is sequence dependent, which may originate from the polarization of the nearby peptide environment.

|
Download:
|
| Fig. 5. TVS within Taylor expansion fitted energy offset and UPS experimental and DTF calculated HOMO and gold Fermi level energy offset of H-1, H-4, H-7 and 7-Ala SAMs molecular junctions. | |
|
|
Table 1 Key electronic structure parameters of 7-Ala, H-1, H-4 and H-7 SAMs on gold substrate. Unit is in eV. |

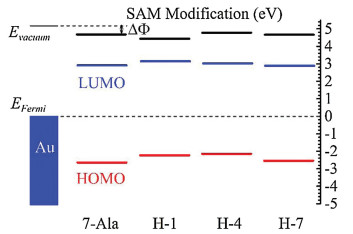
To further approve the doping effect of histidine on the energy landscape and the charge transport, we analyzed the energy state of the studied peptides from UPS results and DFT calculation. The energy offset between HOMO energy levels of four peptides SAMs and gold substrate Fermi energy by UPS measured were illustrated in Fig. 6 and summarized in Table 2. The lowest unoccupied molecular orbital (LUMO) energy level of these peptides, estimated by optical gap from UV measurement, were further away from the gold Feimi energy level than HOMO (~3 eV above gold Fermi level estimeated from the optical bandgap) [33]. Thus HOMO is believed to dominate the charge transport in these peptide SAMs. The doping of histidine into different position of peptdie chain all showed a reduction in peptide HOMO and gold Fermi level energy offset (for H-1, H-4 and H-7 is 2.21, 2.16 and 2.58 eV, respectively and for 7-Ala is 2.67 eV). We can see a position dependence in tuning the energy offset of peptdies H-1, H-4 and H-7 and similar trend has been found from I–V simulation obtained tunneling energy barrier (see Fig. 5 for the comparision in trend).

|
Download:
|
| Fig. 6. Estimated energy diagram of 7-Ala, H-1, H-4 and H-7 SAMs on the gold substrate. | |
|
|
Table 2 Summary of HOMO, LUMO energy levels, energy bandgap and work function of 7-Ala, H-1, H-4 and H-7 SAMs on gold substrate. Unit is in eV. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DFT calculation of the fully stretched peptide molecules was further employed to find the electronic structure information of the four types of peptides. As shown in Fig. 7 (details of the DFT calculation see Supporting information), in presence of histidine, the HOMO energy level was shifted from anchoring sulfor atom to the conjugate imidazole ring of histidine, and their energy levels are much closer to the gold electrode Fermi level (~ 5.1 eV), thus resulting a reduction in effective junction energy barrier. We observe difference in value of energy barrier obtained from UPS measurement, I-V fitting and DFT calculation (Fig. 5). UPS measured increase in peptides HOMO energy (peptides immobilized to gold substrate) compared to DFT calculated HOMO energy (calculated with peptides in vacuum without attachment to electrode), which may originated from the surface-induced gap renormalization effect upon immobilization of the peptide to the gold surface [35]. And further decrease in energy barrier by I-V fitting can be attributed to the interaction of the peptide with the top GaIn electrode. On the other hand, the imidazole HOMO energies did not show a strong sequence dependence. A weak sequence dependent conductance was ever reported on series tunneling in tryptophan doped oligo-alanine junctions [16]. There, the conductance difference was mainly attributed to the contact coupling to the electrode when tryptophan is at C terminal of the peptide relative to in the middle of the peptide. While the TVS analysis of our histidine doped oligo-alanine peptide demonstrated a significant sequence dependent energy barrier. Considering the H-4 peptide took more collapsed conformation assembled to the substrate relative to the other two, the lowered energy barrier might originate from the conformation of H-4. Indeed, in previous study, Sepunaru et al. have found that the transport energy barrier of oligo-alanine in the helix conformation is lower than that of the stretched conformation [15].

|
Download:
|
| Fig. 7. Electronic structure calculation by DFT of H-1, H-4 and H-7 and their comparison with 7-Ala. | |
{kind=link}
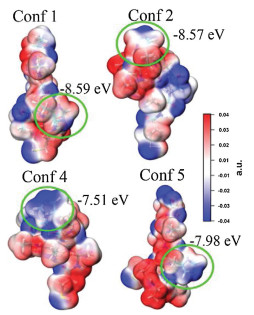
To find the origin of the energy barrier decrease in H-4 peptide, we performed further theoretical calculation. First, we run molecular dynamics to obtain many different conformers of neutral H-4 and picked up those with similar diameters as the monolayer thickness we obtained in the experiment. We then run further DFT calculation on these conformers (see detailed calculation method and results in Supporting information) [36, 37]. We found there exist conformers, of which the HOMO energy levels of the imidazole are higher and closer to the gold electrode Fermi energy levels (HOMO energy levels of Conf 4 and Conf 5 are -7.51 eV and -7.98 eV, respectively). Molecular electrostatic potential surface analysis (Fig. 8) showed that the negative charge group, mainly the carbonyl group of the amide, around the imidazole can significantly increase its negative potentials and HOMO energy levels. We believe this is due to the nitrogen atom with lone pair electron in imidazole side group of H-4, which is quite sensitive to environmental polarity and presents different potential when surrounded by different charged groups. Therefore, the imidazole groups in folded conformers of Conf 4 and Conf 5 in H-4 monolayer may experience an enhancement of the HOMO energy by the carbonyl groups of the amide bond of the peptide chain so that a decreased charge transport barrier.

|
Download:
|
| Fig. 8. Plot of electrostatic surface potential maps of four selected H-4 conformers with imidazole group circled in green solid line. | |
{kind=link}
In summary, we have fabricated molecular junctions by self-assembling peptides, oligo-alanine and histidine doped at different position of oligo-alanine, onto gold substrate as the bottom electrode and using EGaIn as the top electrode. EGaIn measured junction current density revealed series tunneling in these peptide junctions. The average junction current of peptide junctions with histidine doping showed an overall increase compared to oligo-alanine junction current. DFT calculations and UPS measured HOMO and gold Fermi level energy difference suggests that the introducing of histidine into oligo-alanine changes the electronic structure of the peptides, HOMO located on the imidazole side group for H-1, H-4 and H-7 instead of located on sulfur atom for 7-Ala, which reduces the peptide HOMO and gold Fermi level energy difference. This position dependent lowering in peptide HOMO and gold Fermi level energy difference leads to a reduction of the molecular junction effective energy offset, which is supported by TVS within Taylor expansion fitting results. This position dependence may result from the polarity change by histidine doping at different position into the peptide chain. Histidine with effectiveness of mediating charge tunneling and ability to fine tuning molecular junction energy landscape provides to be a promising candidate towards fine tunable functional bio-electronic devices.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 21773169, 21973069, 21805144), Natural Science Foundation of Zhejiang Province (No. LY18B020016), and the PEIYANG Young Scholars Program of Tianjin University (No. 2018XRX-0007). We thank Y. Zou from Institute of Chemistry, Chinese Academy of Sciences for helping with UPS measurement.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.04.013.
| [1] |
M. Kai, K. Takeda, T. Morita, S. Kimura, J. Pept. Sci. 14 (2008) 192-202. |
| [2] |
J.R. Winkler, H.B. Gray, Chem. Rev. 114 (2014) 3369-3380. DOI:10.1021/cr4004715 |
| [3] |
N. Amdursky, D. Marchak, L. Sepunaru, et al., Adv. Mater. 26 (2014) 7142-7161. DOI:10.1002/adma.201402304 |
| [4] |
J. Juhaniewicz, J. Pawlowski, S. Sek, Isr. J. Chem. 55 (2015) 645-660. DOI:10.1002/ijch.201400165 |
| [5] |
C.D. Bostick, S. Mukhopadhyay, I. Pecht, et al., Rep. Prog. Phys. 81 (2018) 026601. DOI:10.1088/1361-6633/aa85f2 |
| [6] |
M. Baghbanzadeh, C.M. Bowers, D. Rappoport, et al., Angew. Chem. Int. Ed. 54 (2015) 14743-14747. DOI:10.1002/anie.201507271 |
| [7] |
L. Venkataraman, J.E. Klare, C. Nuckolls, M.S. Hybertsen, M.L. Steigerwald, Nature 442 (2006) 904-907. DOI:10.1038/nature05037 |
| [8] |
O.E.C. Ocampo, P. Gordiichuk, S. Catarci, et al., J. Am. Chem. Soc. 137 (2015) 8419-8427. DOI:10.1021/jacs.5b01241 |
| [9] |
D.N. Beratan, J.N. Onuchic, J.N. Betts, B.E. Bowler, H.B. Gray, J. Am. Chem. Soc. 112 (1990) 7915-7921. DOI:10.1021/ja00178a011 |
| [10] |
H.B. Gray, B.G. Malmstroem, Biochemistry 28 (1989) 7499-7505. DOI:10.1021/bi00445a001 |
| [11] |
J.N. Onuchic, D.N. Beratan, J.R. Winkler, H.B. Gray, Annu. Rev. Biophys. Biomol. Struct. 21 (1992) 349-377. DOI:10.1146/annurev.bb.21.060192.002025 |
| [12] |
K. Zhou, K. Dai, C. Liu, C. Shen, SmartMat 1 (2020) e1010. |
| [13] |
L. Kong, C. Tang, H.J. Peng, J.Q. Huang, Q. Zhang, SmartMat 1 (2020) e1007. |
| [14] |
X. Chen, Y.Q. Yeoh, Y. He, et al., Angew. Chem. Int. Ed. 59 (2020) 22554-22562. DOI:10.1002/anie.202004701 |
| [15] |
L. Sepunaru, S. Refaely-Abramson, R. Lovrinčic', et al., J. Am. Chem. Soc. 137 (2015) 9617-9626. DOI:10.1021/jacs.5b03933 |
| [16] |
L.A. Zotti, J.C. Cuevas, ACS Omega 3 (2018) 3778-3785. DOI:10.1021/acsomega.7b01917 |
| [17] |
L.A. Zotti, B. Bednarz, J. Hurtado-Gallego, et al., Biomolecules 9 (2019) 580-592. DOI:10.3390/biom9100580 |
| [18] |
C. Guo, X. Yu, S. Refaely-Abramson, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 10785-10790. DOI:10.1073/pnas.1606779113 |
| [19] |
W.M. Schosser, L.A. Zotti, J.C. Cuevas, F. Pauly, J. Chem. Phys. 150 (2019) 7. |
| [20] |
J.R. Winkler, H.B. Gray, J. Am. Chem. Soc. 136 (2014) 2930-2939. DOI:10.1021/ja500215j |
| [21] |
M.R. Kalani, A. Moradi, M. Moradi, E. Tajkhorshid, Biophys. J. 105 (2013) 993-1003. DOI:10.1016/j.bpj.2013.06.047 |
| [22] |
C. Valéry, S. Deville-Foillard, C. Lefebvre, Nat. Commun. 6 (2015) 1-8. |
| [23] |
D. Onidas, J.M. Stachnik, S. Brucker, S. Krätzig, K. Gerwert, Eur. J. Cell Biol. 89 (2010) 983-989. DOI:10.1016/j.ejcb.2010.08.007 |
| [24] |
H.B. Gray, J.R. Winkler, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 3534-3539. DOI:10.1073/pnas.0408029102 |
| [25] |
B. Li, L. Tian, X. He, et al., Phys. Chem. Chem. Phys. 21 (2019) 26058-26065. DOI:10.1039/C9CP04695J |
| [26] |
M.R. Rahman, T. Okajima, T. Ohsaka, Anal. Chem. 82 (2010) 9169-9176. DOI:10.1021/ac101206j |
| [27] |
K.B. Alici, I.F. Gallardo, Sci. Rep. UK. 3 (2013) 1-7. |
| [28] |
R. Schweitzer-Stenner, I. Pecht, C. Guo, J. Phys. Chem. B 123 (2019) 860-868. DOI:10.1021/acs.jpcb.8b09180 |
| [29] |
L. Yuan, L. Jiang, D. Thompson, C.A. Nijhuis, J. Am. Chem. Soc. 136 (2014) 6554-6557. DOI:10.1021/ja5007417 |
| [30] |
P. Rothemund, C.M. Bowers, Z. Suo, G.M. Whitesides, Chem. Mater. 30 (2018) 129-137. DOI:10.1021/acs.chemmater.7b03384 |
| [31] |
L. Jiang, C.S.S. Sangeeth, A. Wan, A. Vilan, C.A. Nijhuis, J. Phys. Chem. C 119 (2015) 960-969. DOI:10.1021/jp511002b |
| [32] |
K.C. Liao, L.Y. Hsu, C.M. Bowers, H. Rabitz, G.M. Whitesides, J. Am. Chem. Soc. 137 (2015) 5948-5954. DOI:10.1021/jacs.5b00448 |
| [33] |
Z. Xie, I. Bâldea, C.E. Smith, Y. Wu, C.D. Frisbie, ACS Nano 9 (2015) 8022-8036. DOI:10.1021/acsnano.5b01629 |
| [34] |
A. Vilan, D. Cahen, E. Kraisler, ACS Nano 7 (2013) 695-706. DOI:10.1021/nn3049686 |
| [35] |
C. Guo, S. Sarkar, S. Refaely-Abramson, et al., Phys. Chem. Chem. Phys. 20 (2018) 6860-6867. DOI:10.1039/C7CP08043C |
| [36] |
S. Grimme, C. Bannwarth, P. Shushkov, J. Chem. Theory Comput. 13 (2017) 1989-2009. DOI:10.1021/acs.jctc.7b00118 |
| [37] |
T. Lu, Molclus Program, Version 1.9.9, http://www.keinsci.com/research/molclus.html.
|