 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055 China;
c Department of Breast and Thyroid Surgery, Shenzhen People's Hospital, Shenzhen 518020, China;
d Department of Student Affairs, Beihang University, Beijing 100191, China
Proximity-induced conjugation on selected residues (e.g., cysteine, lysine or histidine), represents one interesting type of covalent conjugation due to its spontaneity, site-specificity and biocompatibility [1]. In recent years, targeted covalent inhibition has undergone a major revival [2]. Multiple approaches have been developed to selectively conjugate endogenously reactive amino acid residues in proteins [3]. This strategy begins from reversible and noncovalent binding interaction between protein-ligand interaction (ligand-directed (LD) strategy [4]); the binding interaction accurately positions a reactive functional group preinstalled in the ligand to the vicinity of a thiol or ε-amino group nearby the ligand binding site of protein to form a covalent bond. Therefore, the vast majority of covalent inhibitors to date have been rationally designed to target amino acids in close proximity to the ligand site.
Based on the nature of amino acids in proteins, Hamachi and co-workers developed ligand-directed (LD) chemistry to specifically conjugate proteins by labeling targeted residues in living cells [5]. Recently, we also reported a propargylated-sulfonium center triggered thiol-yne coupling for cysteine modification [6]. But the relatively low abundance of cysteines in proteome [7] limits the applicability and diversity of biorthogonal reactions, so other nucleophilic residues, such as lysine have been extensively targeted [8]. Lysine residues contain a potentially nucleophilic primary amine that can be conjugated. The pKa of lysine ε-amino group on the surface of proteins is around 10.4 [9]. Therefore, at physiological pH 7.4, 99.9% of these residues present as protonated species and so are unavailable as nucleophiles. As many enzyme active sites utilize a catalytic lysine residue, so covalent tagging on these lysine residues can thus reveal useful information on the enzyme [10].
MDM4 also known as MDMX, was discovered as a p53-binding protein [11] with a strong similarity to MDM2 in primary amino acid sequence [12]. High overexpression of MDM4 was identified in a wide range of tumor types like sarcomas, carcinomas, melanomas, and leukemias. MDM4 inhibition is likely to be a less hazardous strategy than MDM2 inhibition for restoring p53 function in tumors [13]. Given the importance of MDM4 in p53 reactivation, the development of inhibitors to target MDM4 has been designated a desirable therapeutic goal [14].
In this study, encouraged by the activity of propargylated-sulfonium center in previous reports, we expected that propargylated-sulfonium center could also induce amino-yne reaction on nucleophilic lysine residues and constructed propargylated-sulfonium tethered peptide ligand for MDM4 conjugation model. We first dissolved Boc-Lys-OH, Ac-Cys-OH and propargyl dimethylsulfonium in aqueous ammonium carbonate solutions (pH 8.0). The mixture was stirred at room temperature for 6 h. Unfortunately, we did not find product of sulfonium reacted with lysine in the presence of cysteine (Fig. 1a), possibly because the sulfhydryl group is more nucleophilic than amino group. However, we detected that propargylated sulfonium did react with lysine in a designed model without the presence of free SH. We mixed propargyl dimethylsulfonium and Boc-Lys-OH in water for 12 h and purified the mixture by HPLC to obtain the target product (Fig. 1b).

|
Download:
|
| Fig. 1. (a) Proposed mechanism of thiol-yne reaction with nucleophiles of cysteine residues or amino-yne reaction with nucleophiles of lysine residues. (b) 1H NMR spectrum of 1b product (400MHz in DMSO-d6) after amino-yne reaction. (c) Labelling proteins of wild-type MDM4△met1-asp134 and MD3C△met1-asp134 mutant in the presence or absence of FITC-tagged propargylated sulfonium probe. | |
{kind=link}
To further elucidate the product structure, careful nuclear magnetic resonance (NMR) experiments were performed, including 1H NMR, 13C{1H} NMR [15], heteronuclear singular quantum correlation (HSQC), heteronuclear multiple bond correlation (HMBC), and total correlation spectroscopy (TOCSY) (Fig. 1b and Figs. S1–S6 in Supporting information). According to NMR data and previous report [16], we proposed the product structure shown in Fig. 1b. We envisioned that a proper ligand, better bulky in size, may abate or even inhibit the reactivity of propargylated sulfonium with protein cysteines not in vicinity of the interacting face and make it a suitable warhead to target lysine in proximity. To examine the reactivity of propargylated sulfonium in labelling protein, we first constructed an unselective fluorescein isothiocyanate (FITC)-tagged propargylated-sulfonium probe to react with MDM4△met1-asp134 protein (with one Cys) and a Cys-Ser mutation version (MD3C△met1-asp134). Both of them clearly appeared to be effectively labelled, indicating lysine residues could also be labelled under physiologically relevant conditions (Fig. 1c).
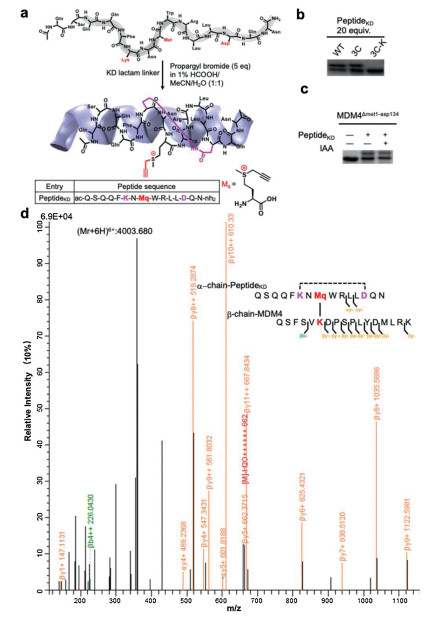
For bulky ligands, we constructed peptide ligand for MDM4△met1-asp134 as shown in Fig. 2a. The SAHp53-8 peptide sequence was chosen based on its extensive studies and clear illustration of its crystal structure (PDB code 3V3B) [17]. MDM4 contains an internal motif, which is structurally analogous to the intramolecular interaction to diminish the binding of MDM4-p53 interaction. Wang et al. reported that a stapled peptide covalently bound to its target protein using sulfonyl fluoride exchange (SuFEx). They improved its inhibition activity on the p53-MDM4 interaction by 10-fold via proximity-enabled bio-reactivity [18]. Here, we incorporated the propargylated sulfonium into the MDM4 binder, SAHp53-8 peptide, to covalently inhibit p53-MDM4 interactions (Scheme 1).

|
Download:
|
| Fig. 2. The propargylated-sulfonium PeptideKD bound MDM4△met1-asp134 protein. (a) Schematic representation of the synthesis of stapled SAHp53-8 Peptides with Lys-Asp (KD) cyclization. (b) Gel shift showing PeptideKD upshifted MDM4△met1-asp134 protein to a much higher molecular weight after conjugation at a 20 equiv. conjugate reaction of PeptideKD with MDM4 wild type and mutants. (d) Conjugated site analysis of PeptideKD with MDM4 at K93 by MS/MS analysis. | |
{kind=link}

|
Download:
|
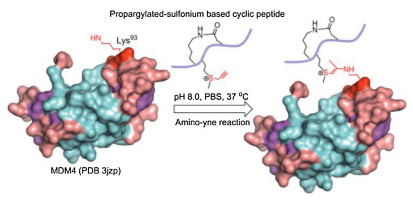
| Scheme 1. The propargylated-sulfonium SAHp53-8 peptide (PeptideKD) bound MDM4△met1−asp134 protein (PDB 3jzp) under PBS solution. | |
{kind=link}
Amino-terminal hydrophobic pocket of MDM4 interacts with p53, thus, we stapled the SAHp53-8 peptide through Lys-Asp (KD) cyclization (Fig. 2a). Therefore, we achieved the cyclic stapled p53 peptides (PeptideKD) to covalently conjugate MDM4△met1-asp134 protein ((PDB 3jzp) [19]) via ligand-directed biorthogonal reaction. We confirmed the reaction of the SAHp53 peptides with MDM4△met1-asp134 protein in pH 8.0 PBS (protein/peptide 20/100 μmol/L, 37 ℃, overnight). As shown in Fig. 2b, the SAHp53 peptides resulted from facile progargylation of thiolethers, successfully bring alkyne group into proximity of the selected lysine site (K93). As a result, the covalent linkage avoids dissociation and effectively stabilizes the inhibition efficiency on p53-MDM4 interaction. Among the SAHp53 peptides, PeptideKD showed better performance in reaction efficiency among three p53 peptide derivatives and showed bonding saturation at 20 equiv. (Fig. 2b). We then tested reaction rate of amino-yne reaction between MDM4 protein and PeptideKD in PBS (pH 8.0) as shown in Fig. S7 (Supporting information) and chose 12 h as reaction time. To further test the conjugated site with PeptideKD, we mutated Lys93 to Arg93 as MDM4 3C-K△met1-asp134 mutant, there is no conjugation between PeptideKD and MDM4 3C-K△met1-asp134 mutant (Fig. 2b).
Next, we tested the site selectivity of PeptideKD with wild type MDM4△met1-asp134 and its mutants. Iodoacetamide (IAA) was used to compete the cysteine residues on protein with PeptideKD. There was no change in labelling with cysteine competitor IAA as shown in Fig. 2c, which further confirmed the lysine reactivity of propargylated sulfonium. Then, we subjected the MDM4–p53 peptide conjugate to proteolysis and analyzed the digested fragments by nano LC–MS/MS. MS analysis of digested fragments confirmed the ligand-directed and lysine-specific complex formation of PeptideKD with MDM4. The symbol "*" in the peptide sequences indicates that the preceding lysine residue reacted with the cross-linking reagent fragmentation spectra of the [QSQQFKNM*WRLLDQN(α) + QSFSVK*DPSPLYDMLRK(β)] cross-linked peptide exported from pLabel v2.4.1 [20]. Some fragments identified are reported on the sequences. A peak at 4003.680Da was observed corresponding to precursor mass (Mr + 6H)6+ of PeptideKD (α-chain) covalently conjugated to MDM4△met1-asp134 (β-chain) (Fig. 2d). It is noted that N-term amine also reacted with our propargylated-sulfonium alkyne. But in this LD strategy, N-term amine, far away from ligand binding site, was not observed reaction trace with alkyne. From the MS evidence we could confirm that propargylated sulfonium could exclusively react with amino group on the lysine residue of protein in proximity-induced reaction.
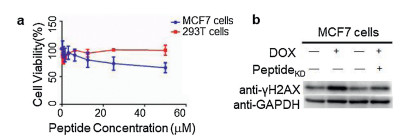
To study whether the PeptideKD could disrupt p53-MDM4 complexes and its regulation on p53 activity, we incubated the p53 peptide with MDM4 in breast cancer cells to measure p53-related DNA damage pathway [21]. The cell viability was first tested at increasing concentration (Fig. 3a). PeptideKD showed low toxicity to MCF-7 cancer cells and 293T cells at 50 μmol/L, consistent with that of propargylated sulfonium was reported [6a]. Then we demonstrated the usefulness of target p53-MDM4 in regulation of DNA-damaged cells. Western blot analysis showed that the level of γH2AX protein was significantly increased in treated MCF7 cells in response to DNA damage induced by 50 μmol/L doxorubicin for 12 h, compared with that of parent cells (Fig. 3b). However, there was only slightly increase in the level of γH2AX protein in MCF7 cells incubated with the PeptideKD for 12 h compared to that that of cells without p53 peptide (Fig. 3b). This phenomenon validated the propargylated-sulfonium p53 peptide as inhibitor of p53-MDM4 binding.

|
Download:
|
| Fig. 3. Effects of propargylated-sulfonium p53 peptide inhibitor on regulation of DNA damage in MCF7 cells. (a) Cell viability of MCF7 cells and 293T cells showed low toxicity of the propargylated-sulfonium p53 peptide. Error bars are shown as mean±SEM of at least two independent measurements. (b) The propargylated-sulfonium p53 PeptideKD inhibit p53-MDM4 binding in cancer cells. MCF7 cells were incubated with 50 μmol/L peptide or equivalent volume of DMSO for overnight, and the level of γH2AX was determined in protein in vivo by Western blotting. | |
{kind=link}
In conclusion, we showed an amino-yne type reaction triggered by propargylated sulfonium center and a lysine in vicinity and developed a propargylated sulfonium tethered p53 peptide (PeptideKD) that can specifically conjugate lysine of MDM4△met1-asp134 protein upon proximity-induced biorthogonal reaction. The propargylated sulfonium p53 peptide can be considered as MDM4 inhibitor to prevent p53-MDM4 binding, which forms the basis for the therapeutic impact of MDM4 targeting. With further undergoing studies, we believed that the new propargylated sulfonium-strategy could be optimized in other residues' modification and be extended in more biological applications, such as MDM4 inhibitor design and antibody conjugated drug development.
Declaration of competing interestThe authors declare that they have no known competing financial interests or persona; relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge financial support from the National Key Research and Development Program "Synthetic Biology" Key Special Project of China (No. 2018YFA0902504); the Natural Science Foundation of China (Nos. 21778009 and 21977010); the Natural Science Foundation of Guangdong Province (Nos. 2020A1515010766, 2020A1515010522, 2019A1515111184 and 2019A1515110489); the Shenzhen Science and Technology Innovation Committee (Nos. JCYJ20180507181527112, JCYJ20180508152213145, and JCYJ20170817172023838). We acknowledge financial support from Beijing National Laboratory of Molecular Science Open Grant (No. BNLMS20160112) and Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (No. 2019SHIBS0004). This work is supported by Proteomic Platform of Pingshan Translational Medicine Center, Shenzhen Bay Laboratory, and the high-performance computing platform of Peking University.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.04.015.
| [1] |
S.S. Gallagher, J.E. Sable, M.P. Sheetz, V.W. Cornish, ACS Chem. Biol. 4 (2009) 547-556. DOI:10.1021/cb900062k |
| [2] |
(a) T.A. Baillie, Angew. Chem. Int. Ed. 55 (2016) 13408-13421. (b) J. Singh, R.C. Petter, T.A. Baillie, A. Whitty, Nat. Rev. Drug Discov. 10 (2011) 307-317. |
| [3] |
B.F. Cravatt, E.J. Sorensen, Curr. Opin. Chem. Biol. 4 (2000) 663-668. DOI:10.1016/S1367-5931(00)00147-2 |
| [4] |
K. Amaike, T. Tamura, I. Hamachi, Chem.Commun.(Camb.) 53 (2017) 11983. |
| [5] |
(a) H. Nonaka, S.H. Fujishima, S.H. Uchinomiya, A. Ojida, I. Hamachi, J. Am. Chem. Soc. 132 (2010) 9301-9309. (b) H. Nonaka, S. Tsukiji, A. Ojida, I. Hamachi, J. Am. Chem. Soc. 129 (2007) 15777-15779. |
| [6] |
(a) Z. Hou, D. Wang, Y. Li, et al., J. Org. Chem. 85 (2020) 1698-1705. (b) D. Wang, M. Yu, N. Liu, et al., Chem. Sci. 10 (2019) 4966-4972. |
| [7] |
S.M. Marino, V.N. Gladyshev, J. Mol, Biol. 404 (2010) 916. |
| [8] |
D.A. Shannon, E. Weerapana, Curr. Opin. Chem. Biol 24 (2015) 18-26. DOI:10.1016/j.cbpa.2014.10.021 |
| [9] |
S. De, A.C. Chan, H.J. Coyne 3rd, J. Mol. Biol. 426 (2014) 1390-1406. DOI:10.1016/j.jmb.2013.11.031 |
| [10] |
A.C. Carrera, K. Alexandrov, T.M. Roberts, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 442-446. DOI:10.1073/pnas.90.2.442 |
| [11] |
S. Francoz, P. Froment, S. Bogaerts, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 3232-3237. DOI:10.1073/pnas.0508476103 |
| [12] |
A. Shvarts, W.T. Steegenga, N. Riteco, EMBO J. 15 (1996) 5349-5357. DOI:10.1002/j.1460-2075.1996.tb00919.x |
| [13] |
F. Bernal, M. Wade, M. Godes, Cancer Cell 18 (2010) 411-422. DOI:10.1016/j.ccr.2010.10.024 |
| [14] |
(a) K. Itahana, H. Mao, A. Jin, et al., Cancer Cell 12 (2007) 355-366. (b) B.L. Grasberger, T. Lu, C. Schubert, et al., J. Med. Chem. 48 (2005) 909-912. |
| [15] |
C. Qian, M.M. Zhou, Cell Mol. Life Sci. 63 (2006) 2755-2763. DOI:10.1007/s00018-006-6274-5 |
| [16] |
T. Sugita, M. Eida, H. Ito, J. Org. Chem. 52 (1987) 3789-3793. DOI:10.1021/jo00226a012 |
| [17] |
G.M. Popowicz, A. Czarna, T.A. Holak, Cell Cycle 7 (2008) 2441-2443. DOI:10.4161/cc.6365 |
| [18] |
C. Hoppmann, L. Wang, Chem. Commun 52 (2016) 5140-5143. DOI:10.1039/C6CC01226D |
| [19] |
J. Phan, Z. Li, A. Kasprzak, J. Biol. Chem. 285 (2010) 2174-2183. DOI:10.1074/jbc.M109.073056 |
| [20] |
(a) L.H. Wang, D.Q. Li, Y. Fu, et al., Rapid Commun. Mass Spectrom. 21 (2007) 2985-2991; (b) D. Li, Y. Fu, R. Sun, et al., Bioinformatics 21 (2005) 3049-3050. |
| [21] |
B. Vogelstein, D. Lane, A.J. Levine, Nature 408 (2000) 307-310. DOI:10.1038/35042675 |