 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory of Crystal Materials, Shandong University, Ji'nan 250100, China
The multifunctional catalysts that combine different active sites can endow the whole catalytic process with improved efficiency in terms of economy, environment and energy consumption [1-4]. Currently, the most effective method of constructing advanced multifunctional catalysts is atomic layer deposition (ALD). ALD is extremely dependent on its unique sequential self-limiting surface reactions, thereby being capable of dispersing different functional centers with high spatial uniformity [5,6]. However, the use of ALD methods has problems, such as expensive equipment and precursors and complex operation. If the spatial separation of multiple sites can be achieved by a simple impregnation method, it will be of great importance for the design and development of industrial catalyst. Unfortunately, the commonly used one-pot or stepwise impregnation method definitely result in either an alloy or an interfacial structure according to the classical synthetic chemistry theory of materials [7-9], thus encountering significant limitations in the spatial separation of different active sites. Therefore, spatial separation of complementary functional sites at the nanoscale in multifunctional catalysts remains a major challenge aiming at steering reaction sequence and paths.
Gold-titania (Au/TiO2) nanocatalyst has received special interest in photocatalytic chemistry for solar light-induced hydrogen/hydrocarbon production because of their attractive electronic, optical properties, and excellent structural stability [10-19]. When Au/TiO2 photocatalyst is irradiated by UV, Au can function as an electron sink to execute the hydrogen evolution half-reaction by accumulating the photo-generated electrons in conduction band of the light-absorbing TiO2. The photo-generated holes will diffuse to the surface of TiO2 to complete the oxygen evolution half-reaction, thereby promoting the separation and lifetime of electron-hole pair. However, the remaining high-concentrated and highly active holes trapped in TiO2 still have to migrate to the surface to participate in the reaction and suffer from a high chance of recombining with electrons [20,21]. Although the surface plasmon resonance (SPR) effect of Au particles is capable of initiating hydrogen evolution exclusively with visible light irradiating, only a small percentage of UV light in real sun light have been proved to lead to a significant improvement in terms of hydrogen production amount for Au/TiO2 photocatalysts. Therefore, simultaneously manipulating the separation of photo-generated electron-hole pair through a dual co-catalyst stabilizing is of great importance to achieve a high photocatalytic efficiency of Au/TiO2 for the water splitting under UV-irradiation. Moreover, the plasmonic property of Au can induce photocatalytic activity of TiO2 under visible-light illumination through hot electron generation and injection, simultaneously resulting in the creation of holes in Au [22,23]. As the hot electrons are injected into conduction band of semiconductor, the Schottky barrier is formed at the junction between Au and TiO2 and has a negative impact on the photocatalytic activity by hindering the continued injection of hot electrons [24-26]. The manipulation of the height of Schottky barrier is of great importance in promoting the photocatalysis efficiency of Au/TiO2 under visible-light illumination through accelerating the injection of plasmon-induced electron from Au to semiconductor. Unfortunately, the development of Au/TiO2-based systems has been on the horns of a dilemma in addressing the above key scientific issues because of lacking elaborate structural design [27]. Therefore, the precise control of different functional centers on the nanosurface of semiconductor is of great importance in promoting photocatalytic efficiency of Au/TiO2 under both UV and visible band of solar light by highly efficient separation and transport of the photo-generated electrons and holes, which is a significant scientific and technological challenge.
Herein, we reported the spatial separation of dual-site Au and RuO2 on the nanosurface of TiO2 (Au/TiO2/RuO2) via facile impregnation method by referring to the concept of unique sequential self-limiting surface reactions. The strong metal-support interaction (SMSI) and the lattice matching (LM) can induce the encapsulation of Au nanoparticles by an impermeable TiOx overlayer and increase the stability of RuO2/TiO2 interface, thereby preventing the aggregation of dual-site Au and RuO2. The resultant Au/TiO2/RuO2 exhibits a great enhancement for photocatalytic hydrogen evolution under solar light irradiation without sacrificial agents. The promoted separation efficiency of the photo-generated electron and hole pairs and the lowering height of the Schottky barrier in Au/TiO2/RuO2 through the engineering of spatially separated dual-site Au and RuO2 are suggested to be responsible for the robust photocatalytic performance.
Synthesis of Au/TiO2-S: 1 g of TiO2 (Degussa P25) was firstly added to 40 mL of HAuCl4·4H2O aqueous solution (6.3 mmol/L), and then the pH of the mixture was adjusted to pH 9.0 with 0.1 mol/L NaOH. The slurry was magnetically stirred for 1 h at 65 ℃. The solid powder was collected after washing with ultrapure water for several times. The Au/TiO2 is obtained after an overnight dryness at 60 ℃ and further calcination at 250 ℃ for 2 h in air.
Synthesis of Au@TiO2/TiO2: An impermeable TiOx overlayer is wrapped on the surface of Au (Au@TiO2/TiO2) through the strong metal-support interaction (SMSI) between Au and TiO2 after a treatment with mixed gas of 10 vol% H2/He at 500 ℃ for 1 h at a heating rate of 2 ℃/min.
Synthesis of Au/TiO2: Au/TiO2 was prepared through removing the TiOx overlayer at 400 ℃ in 10% O2/He for 1 h at a heating rate of 2 ℃/min.
Synthesis of Au/RuO2/TiO2-m: Au/RuO2/TiO2-m was prepared by mixing the Au and Ru precursors at the beginning. All other steps are the same with the synthetic process of Au/TiO2.
Synthesis of Au/RuO2/TiO2-c: Au/RuO2/TiO2-c was prepared without the SMSI process, i.e., the step for preparing Au@TiO2/TiO2.
Synthesis of Au/TiO2/RuO2: As for the synthesis of Au/TiO2/RuO2, the Ru precursor (RuCl3) was mixed with Au@TiO2/TiO2. All other steps are the same with the synthetic process of Au/TiO2.
X-ray diffraction (XRD) patterns were recorded on a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å). Scanning electron microscopy (SEM) images were obtained with a Hitachi Regulus-8100 Field Emission Scanning Electron Microscope. Transmission electron microscopy (TEM) images were achieved by using a Hitachi H-7700 TEM operating at 100 kV. High-resolution transmission electron microscopy (HRTEM) was conducted on a FEI Tecnai G2 F20 STwin microscope at 200 kV. X-ray photoelectron spectroscopy (XPS) signals were collected by a Thermo Fisher ESCALAB 250Xi spectrometer applying monochromatic Al Kα X-ray sources (1486.6 eV) at 2.0 kV and 20 mA. UV–vis diffuse reflectance spectra (DRS) were recorded on a UV–vis spectrophotometer (UV-3600, Shimadzu). Raman measurements were conducted on the LabRam HR Evolution system. The PL spectra was obtained with a Horiba Luminescence Spectrometer with 375 nm as a laser excitation. The time-resolved PL lifetime was achieved on FLS980, Edinburgh with 375 nm as a laser excitation.
The photocatalytic activity of samples for hydrogen evolution was evaluated by applying a photocatalytic hydrogen evolution equipment (PerfectLight, Beijing Co., Ltd.). Typically, 0.1 g of catalyst was uniformly dispersed in 100 mL pure water with ultrasonication for 5 min but without using any sacrifice reagents like methanol. The simulated solar is obtained from Xe lamp with AM 1.5 filter, and a uniform power intensity of 100 mW/cm2 was calibrated. A homeothermic cooling circulation system was applied to keep the test temperature of 15 ℃. The online gas chromatography (GC) equipped with a TCD detector was used to quantify the cumulative amount of hydrogen for every 1 h. Photocurrent generation was measured using a three-electrode system. The reference and counter electrodes are saturated calomel electrode (SCE) and Pt sheet, respectively. The Na2SO4 aqueous solution (50 mL, 0.5 mol/L, pH 6.8) was used as electrolyte.
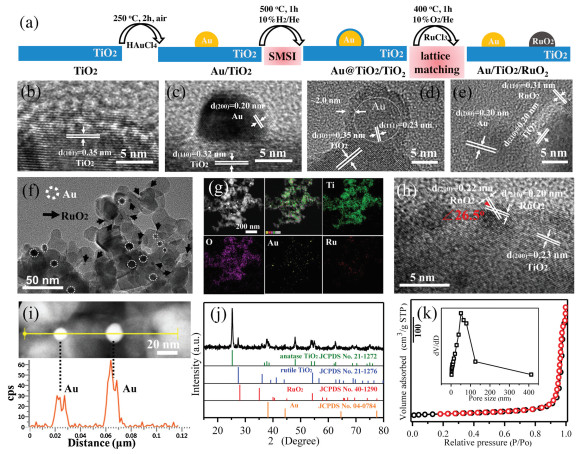
The preparation process of Au/TiO2/RuO2 with spatially separated dual-site Au and RuO2 is schematically illustrated in Fig. 1a. The sample of bare Au nanoparticles on TiO2 (Au/TiO2) was firstly prepared starting from the pristine TiO2 through impregnation-precipitation method and followed by a calcination at 250 ℃ for 2 h in air. The strong metal-support interaction (SMSI) between Au and TiO2 triggered the formation of an impermeable TiOx overlayer on the surface of Au (Au@TiO2/TiO2) after treatment with mixed gas of 10 vol% H2/He at 500 ℃ for 1 h [28]. The pre-formed TiOx overlayer can function as a physical separation barrier to the permeation of the second precursor. And then the RuCl3 was added as precursor of RuO2. When treated with oxidation condition of 10 vol% O2/He at 400 ℃ for 1 h, the TiOx overlayer retreats. Simultaneously, the Au nanoparticle is exposed and the spatially separated RuO2 is formed. The lattice matching between RuO2 and TiO2 can increase the stability of RuO2/TiO2 interface and thus prevent the aggregation of dual-site Au and RuO2 in the oxidation calcination process. The structure evolution from TiO2, Au/TiO2, Au@TiO2/TiO2 to Au/TiO2/RuO2 is exhibited in Figs. 1b–e in the corresponding HRTEM images. Fig. 1b shows clean nanosurface of pristine TiO2. The Au nanoparticle with a size of about 10 nm is formed on the surface of TiO2 according to the analysis of lattice spacing (Fig. 1c). A uniform coating with a thickness of approximately 2 nm is clearly observed on the surface of Au nanoparticle, implying the formation of SMSI-induced TiOx overlayer in Au@TiO2/TiO2 (Fig. 1d). In addition, the unnoticeable size change of Au nanoparticle from Au/TiO2 to Au@TiO2/TiO2 with a high temperature calcination treatment indicates the SMSI-induced robust sintering-resistant ability of Au nanocatalyst. As shown in Fig. 1e, the lattice spacing of 0.20, 0.31 and 0.20 nm can be attributed to the (200) plane of Au, (110) plane of RuO2, and (210) plane of TiO2, respectively, signifying the co-existence of spatially separated Au and RuO2 nanoparticles on the nanosurface of TiO2. The RuO2 nanoparticle is evaluated to be about 3 nm. The spatially distance between dual-site Au and RuO2 is about 10 nm, which ensures the spatial separation of the photo-generated electron and hole pairs on TiO2. No visible changes in the morphology are observed between the pristine P25-TiO2 and the resultant Au/TiO2/RuO2 (Fig. S1 in Supporting information). The low-magnification TEM image (Fig. 1f) indicate the high dispersion degree of dual-site Au and RuO2 on the surface of TiO2. The EDX elemental mapping (Fig. 1g) of Au/TiO2/RuO2 demonstrates the uniform distribution of Ti, O, Au and Ru elements throughout the sample, further implying the good spatial conformity. The contents of Au and Ru in Au/TiO2/RuO2 are roughly evaluated to be 1.4 wt% and 1.9 wt% based on the EDX result (Fig. S2 in Supporting information). In order to give a stronger evidence about the distribution of Au and RuO2, a more detailed analysis of TiO2/RuO2 and Au/TiO2 interfaces is provided. As show in Fig. 1h, the two crystalline planes of the small nanoparticle with lattice spacing of 0.22 and 0.20 nm, respectively, and an angle of 26.5° are attributed to the features of (200) and (210) planes of RuO2, verifying that the small nanoparticle is RuO2 functional site. Importantly, the current result presents the lattice mismatch between RuO2 and TiO2 is only about 4%. The < 5% of lattice mismatch is normally suggested to provide a highly stabilized TiO2/RuO2 interface [29]. Moreover, the RuO2 and rutile TiO2 in P25 have the same tetragonal crystalline structure and specially their lattice mismatch along [101] direction is only 2.2% [30]. The above analysis signifies the presence of high-level lattice match between RuO2 and TiO2, which is capable of preventing the aggregation of RuO2 and Au dual sites during calcination process. A line scanning of big nanoparticles in Au/TiO2/RuO2 (Fig. 1i) is conducted and the positions of Au signal peaks are well matched with the distance of the two nanoparticles, thereby proving that the bigger nanoparticle is Au nanoparticles. Moreover, as exhibited in Fig. 1j, XRD pattern of Au/TiO2/RuO2 demonstrate the existence of anatase (JCPDS No. 21-1272) and rutile (JCPDS No. 21-1276) TiO2 from P25, and Au (JCPDS No. 04-0784) and RuO2 (JCPDS No. 40-1290) phases. Note that the phases of Au and RuO2 are isolated with each other, further verifying the spatial separation of Au and RuO2 in Au/TiO2/RuO2. The nitrogen physisorption isotherm of Au/TiO2/RuO2 (Fig. 1k) indicates a high specific surface area (SBET) of 43 m2/g and the most probable pore size of about 50 nm (inset). Compared with 50 m2/g of the pristine P25-TiO2 (Fig. S3 in Supporting information), the result indicates the loading of Au and RuO2 nanoparticles. Taken together, the spatially separated dual-site Au and RuO2 on the nanosurface of TiO2 (Au/TiO2/RuO2) is successfully constructed through programmed strong metal-support interaction (SMSI) and lattice matching (LM).

|
Download:
|
| Fig. 1. (a) Schematic illustration of the preparation process of Au/TiO2/RuO2. The corresponding HRTEM images to the preparation process from (b) TiO2, (c) Au/TiO2, (d) Au@TiO2/TiO2 (e) to Au/TiO2/RuO2. (f) HRTEM image of Au/TiO2/RuO2. (g) EDX elemental mapping. (h, i) HRTEM images and line scanning of Au/TiO2/RuO2. (j) XRD pattern. (k) N2 physisorption isotherm of Au/TiO2/RuO2. | |
{kind=link}
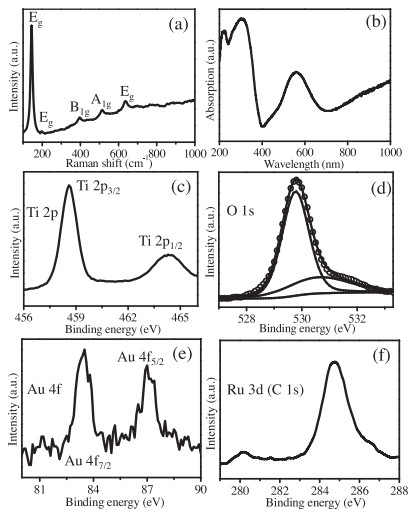
Raman spectroscopy is a highly sensitive analytical method for crystallinity and microstructures of the materials. The Raman spectra of Au/TiO2/RuO2 and P25-TiO2 is shown in Fig. 2a and Fig. S4 (Supporting information), respectively. The well-resolved peaks at 146, 198, 399, 516 and 640 cm−1 are attributed to the characteristic peaks of Eg, Eg, B1g, A1g and Eg in TiO2 [31]. The optical property of Au/TiO2/RuO2 is studied with UV–vis diffuse reflectance spectrum and exhibited in Fig. 2b. The absorption in the UV region is attributed to the band transition of TiO2 [32]. The absorption in the visible region centered at 560 nm is arising from surface plasmon resonance of Au nanoparticles. The uplifted tail after 700 nm should be ascribed to the transition of RuO2 band [33]. Fig. 2c shows two XPS signals of Ti 2p3/2 and Ti 2p1/2 levels centered at about 458.6 and 464.3 eV in the Ti 2p spectrum [34,35]. The O 1s signal in Fig. 2d can be fitted into two peaks centered at about 529.7 and 530.6 eV attributed to the lattice oxygen and the adsorbed oxygen species, respectively [31,33,36]. The existence of two peaks at 83.4 eV for Au 4f7/2 and 87.0 eV for Au 4f5/2 in Au 4f spectroscopy (Fig. 2e) suggest the purely metallic state Au species [37]. The XPS spectroscopy of Ru 3d3/2 (Fig. 2f) is overlapped with that of C 1s at 284.8 eV. The signal peak at a binding energy of 280.2 eV of Ru 3d5/2 demonstrates the presence of Ru4+ species in RuO2 [33,38]. These results further confirmed the co-existence of Au and RuO2 in the resultant Au/TiO2/RuO2.

|
Download:
|
| Fig. 2. (a) Raman spectroscopy, (b) UV–vis diffuse reflectance spectrum, XPS results of (c) Ti 2p, (d) O1s, (e) Au 4f and (f) Ru 3d (and C 1s) of Au/TiO2/RuO2. | |
{kind=link}
The photocatalytic performances of Au/TiO2/RuO2 and the reference samples were investigated for water splitting under solar light irradiation without methanol as sacrificial reagent. As given in Fig. 3a, the amount of hydrogen production is unnoticeable for the pristine P25 without any co-catalyst even after 6 h irradiation. However, Au@TiO2/TiO2 that has a TiOx overlayer on Au nanoparticle gives a promoted photocatalytic hydrogen production of 127 μmol/g for 6 h, which should be attributed to the separation of photo-generated carriers and the surface plasmon resonance (SPR) of Au nanoparticles. By contrast, Au/TiO2 with exposed Au nanoparticle shows an obvious improvement in photocatalytic activity, and the hydrogen production can reach to 203 μmol/g for 6 h. Unfortunately, the Au/RuO2/TiO2 samples prepared through either mixing the Au and Ru precursors (Au/RuO2/TiO2-m) or canceling the SMSI process (Au/RuO2/TiO2-c) present a slight activity increase compared with Au@TiO2/TiO2. The performance of 157 and 183 μmol/g for 6 h of Au/RuO2/TiO2-m/-c is still lower than 203 μmol/g of the Au/TiO2 with exposed Au nanoparticle. The activity differences between Au@TiO2/TiO2, Au/RuO2/TiO2-m/-c and Au/TiO2 should be ascribed to the reason that the TiOx overlayer on Au nanoparticle in Au@TiO2/TiO2 and the non-separated Au and RuO2 dual sites could weaken the functions of Au as co-catalyst and SPR site. Interestingly, the Au/TiO2/RuO2 with spatial separated Au and RuO2 exhibits the highest amount of hydrogen production as high as 504 μmol/g. The calculated rates of hydrogen production (Fig. 3b) of P25, Au@TiO2/TiO2, Au/RuO2/TiO2-m, Au/RuO2/TiO2-c, and Au/TiO2/RuO2 are 0, 21.3, 26.2, 30.5, 33.8 and 84.0 μmol h−1 g−1. The performance of Au/TiO2/RuO2 is 2.5 times higher than the reference Au/TiO2 and non-separated Au/RuO2/TiO2 samples. Moreover, as shown in Fig. 3c, a long-term photocatalytic test of Au/TiO2/RuO2 demonstrate a stable line pattern of hydrogen production, suggesting its excellent stability. Therefore, the Au/RuO2/TiO2 has exhibited attractive photocatalytic activity and preferable long-term stability in hydrogen evolution under solar light irradiation without methanol as sacrificial reagent. In order to unveil the potential fate of light-generated electron-hole pairs, the charge carrier recombination processes of Au/TiO2 and Au/TiO2/RuO2 are analyzed by photocurrent response, photoluminescence (PL) emission and PL lifetime. As shown in Fig. 3d, the photocurrent of Au/TiO2/RuO2 is higher than that of Au/TiO2. The intensity of the PL spectra can signify the recombination amount between electron and holes under emission of photons [33,39]. Compared with Au/TiO2, Au/TiO2/RuO2 exhibits lower intensity of PL (Fig. 3e). Time-resolved PL measurement (Fig. 3f) exhibits that the intensity of Au/TiO2/RuO2 decays much more slowly than that of Au/TiO2, verifying a longer lifetime of photo-generated elector and hole pairs in Au/TiO2/RuO2 [40,41]. The above results confirmed that the spatially separated Au and RuO2 in Au/TiO2/RuO2 can greatly enhance the separation and transport effciency of photo-generated electron-hole pairs, thereby leading to an attractive promotion for water splitting under solar light irradiation without methanol as sacrificial reagent.

|
Download:
|
| Fig. 3. (a, b) Photocatalytic performances. (c) Cycling stability. (d) Photocurrent response. (e) Photoluminescence (PL) emission. (f) Time-resolved PL measurement. | |
{kind=link}
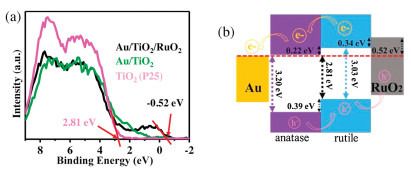
In addition, X-ray photoelectron (XPS) valence band spectra of P25, Au/TiO2 and Au/TiO2/RuO2 were characterized in Fig. 4a to further disclose the changes in electronic structures. The valence band edge of P25 is located at approximately 2.81 eV relative to Fermi level, which is attributed to the top level of rutile-TiO2 valence band arising from the band alignment of anatase-TiO2 and rutile-TiO2 [30,42]. The rutile-TiO2 features a band gap of 3.03 eV with Fermi level lying within the band range, and its bottom level of conduction band is 0.22 eV above Fermi level [43]. The anatase-TiO2 has a band gap of 3.20 eV with Fermi level locating right at its conduction band bottom, and its top level of valence band is about 0.39 lower than that of rutile-TiO2. As shown in the spectrum of Au/TiO2/RuO2, the band edge of RuO2 is approximately −0.52 eV [30]. The result implies that the Fermi level should be lying within its states, but its high-density states are still locating under Fermi level. The level is obviously higher than the valence band of rutile-TiO2, thereby facilitating the separation and transport of electrons from RuO2 to rutile-TiO2 or holes from rutile-TiO2 to RuO2 across their interfaces. Taken together, a full description about the separation and transport of photo-generated electron-hole pairs in Au/TiO2/RuO2 is given as follows. When the P25-TiO2 is irradiated with UV light, the photo-generated electron in conduction band of rutile-TiO2 flows to conduction band of anatase-TiO2. While the holes go in the opposite direction from valence band of anatase-TiO2 to that of rutile-TiO2. The electrons accumulated in anatase-TiO2 side could transfer to Au nanoparticle to trigger hydrogen evolution. The holes accumulated in rutile-TiO2 side can further migrate to RuO2 nanoparticle, thereby preventing the recombination of photo-generated electron-hole pairs. Therefore, a tandem charge transfer with spatially separated Au and RuO2 in Au/TiO2/RuO2 (Fig. 4b) can further enhance the separation and transport efficiency of photo-generated electron-hole pairs in TiO2-based photocatalysts and thus promote their photochemical performances [44-46]. As for visible-light illumination, hot electrons generated from the plasmonic property of Au nanoparticle can transfer to TiO2 to participate in photocatalytic hydrogen evolution, and then the Schottky barrier is formed at the junction between Au and TiO2 [47,48]. The height of Schottky barrier is of great importance in promoting the photocatalysis efficiency of Au/TiO2 under visible-light illumination through facilitating the injection of plasmon-induced electron from Au to semiconductor. The valence band maxima of Au/TiO2 and Au/TiO2/RuO2 shift to lower binding energies by 0.33 and 0.23 eV, respectively, in comparison to pristine TiO2, which should correspond to the Schottky barrier height between Au and TiO2. The surface plasmon resonance of Au nanoparticles can be triggered at about 550 nm of visible light. The energy of generated electrons on the surface of Au nanoparticles is approximately 2 eV, which is sufficient to flow across 0.33 in Au/TiO2 and 0.23 eV in Au/TiO2/RuO2 of Schottky barrier height to the TiO2 (Fig. S5 in Supporting information) [24]. Importantly, the lower Schottky barrier height in Au/TiO2/RuO2 means that lower energy is needed to induce the transfer of hot electrons from Au to TiO2 in comparison with Au/TiO2, which is more conducive to photocatalytic activity under visible light irradiation. Therefore, the spatially separated dual-site Au and RuO2 on the nanosurface of TiO2 can effectively separate the photo-generated carriers and lower the height of the Schottky barrier, respectively, under UV and visible light irradiation, thus promoting the photocatalytic performance.

|
Download:
|
| Fig. 4. (a) X-ray photoelectron (XPS) valence band spectra. (b) Band alignment of Au/TiO2/RuO2. | |
{kind=link}
In conclusion, we successfully constructed spatially separated dual-site Au and RuO2 on the nanosurface of TiO2 (Au/TiO2/RuO2) through the strong metal-support interaction (SMSI) and the lattice matching (LM). The resultant Au/TiO2/RuO2 exhibits robust photocatalytic hydrogen evolution as high as 84 μmolh−1 g−1 under solar light irradiation without sacrificial agents, which is 2.5 times higher than the reference Au/TiO2 and non-separated Au/RuO2/TiO2 samples. The spatially separated dual-site Au and RuO2 on the nanosurface of TiO2 can effectively separate the photo-generated carriers through a tandem charge transfer and lower the height of the Schottky barrier through facilitating hot electron migration, respectively, under UV and visible light irradiation. The work provides an attractive approach for spatially separating different function sites by programmed SMSI and LM states to refine catalytic performance.
Declaration of competing interestThe authors claim that no conflict of interest exists in the submission of this manuscript, and manuscript is approved by all authors for publication. I would like to declare on behalf of my coauthors that the work described is original research and has not been published previously, which is not under consideration for publication elsewhere in whole or in part.
AcknowledgmentsThis work was supported by the National Key Research and Development Program of China (No. 2017YFB0405400), Shandong Provincial Natural Science Foundation (Nos. ZR2019BB025 and ZR2018ZC0842), the Project of "20 items of University" of Jinan (No. 2018GXRC031).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.04.012.
| [1] |
T.L. Lohr, T.J. Marks, Nat. Chem. 7 (2015) 477-482. DOI:10.1038/nchem.2262 |
| [2] |
H. Wang, X. Zhang, J. Wang, et al., Sci. China Mater. 63 (2020) 1054-1064. DOI:10.1007/s40843-019-1268-7 |
| [3] |
Q. Han, Z. Cheng, J. Gao, et al., Adv. Funct. Mater. 27 (2017) 1606352. DOI:10.1002/adfm.201606352 |
| [4] |
L. Chen, Y. Wang, C. Wu, et al., Nanoscale 12 (2020) 13484-13490. DOI:10.1039/D0NR02556A |
| [5] |
B. Zhang, Y. Qin, ACS Catal. 8 (2018) 10064-10081. DOI:10.1021/acscatal.8b02659 |
| [6] |
Y. Wang, D. Wang, Y. Li, SmartMat 2 (2021) 56-75. DOI:10.1002/smm2.1023 |
| [7] |
S. Shen, X. Wang, Chem. Commun. 46 (2010) 6891-6899. DOI:10.1039/c0cc00900h |
| [8] |
C.Y. Wang, C.H. Yang, Z.C. Zhang, Rare Met. 39 (2020) 1353-1355. DOI:10.1007/s12598-020-01568-1 |
| [9] |
C.H. Yang, F. Nosheen, Z.C. Zhang, Rare Met. 40 (2021) 1412-1430. DOI:10.1007/s12598-020-01600-4 |
| [10] |
D.A. Panayotov, A.I. Frenkel, J.R. Morris, ACS Energy Lett. 2 (2017) 1223-1231. DOI:10.1021/acsenergylett.7b00189 |
| [11] |
Y. Gao, W. Nie, Q. Zhu, et al., Angew. Chem. Int. Ed. 59 (2020) 18218-18223. DOI:10.1002/anie.202007706 |
| [12] |
J. Wan, W. Chen, C. Jia, et al., Adv. Mater. 30 (2018) 1705369. DOI:10.1002/adma.201705369 |
| [13] |
X. Wang, F. Wang, Y. Sang, H. Liu, Adv. Energy Mater. 7 (2017) 1700473. DOI:10.1002/aenm.201700473 |
| [14] |
C. Dong, J. Ji, Z. Yang, et al., Chin. Chem. Lett. 30 (2019) 853-862. DOI:10.1016/j.cclet.2019.03.020 |
| [15] |
Q. Guo, C. Zhou, Z. Ma, X. Yang, Adv. Mater. 31 (2019) 1901997. DOI:10.1002/adma.201901997 |
| [16] |
G. Centi, SmartMat 1 (2020) e1005. |
| [17] |
Y. Lu, W.J. Yin, K.L. Peng, et al., Nat. Commun. 9 (2018) 2752. DOI:10.1038/s41467-018-05144-1 |
| [18] |
L. Zeng, C. Xue, Nano Res. 14 (2021) 934-944. DOI:10.1007/s12274-020-3099-8 |
| [19] |
Q. Wei, D.L. Kuhn, Z. Zander, et al., Nano Res. 14 (2021) 3228-3233. DOI:10.1007/s12274-021-3427-7 |
| [20] |
S. Wang, B. Zeng, C. Li, Chin. J. Catal. 39 (2018) 1219-1227. DOI:10.1016/S1872-2067(18)63094-3 |
| [21] |
Q. Han, Z. Cheng, B. Wang, et al., ACS Nano 12 (2018) 5221-5227. DOI:10.1021/acsnano.7b08100 |
| [22] |
S. Linic, S. Chavez, R. Elias, Nat. Mater. 20 (2021) 916-924. DOI:10.1038/s41563-020-00858-4 |
| [23] |
X. Li, H.O. Everitt, J. Liu, Nano Res. 13 (2020) 1268-1280. DOI:10.1007/s12274-020-2694-z |
| [24] |
G. Žerjav, J. Zavašnik, J. Kovač, A. Pintar, Appl. Surf. Sci. 543 (2021) 148799. DOI:10.1016/j.apsusc.2020.148799 |
| [25] |
N. Jiang, D. Li, L. Liang, et al., Nano Res. 13 (2020) 1354-C1362. DOI:10.1007/s12274-019-2599-x |
| [26] |
Z. Wang, R. Li, C. Su, K.P. Loh, SmartMat 1 (2020) e1013. |
| [27] |
B. Ni, H. Cölfen, SmartMat 2 (2021) 17-32. DOI:10.1002/smm2.1021 |
| [28] |
S. Liu, W. Xu, Y. Niu, et al., Nat. Commun. 10 (2019) 5790. DOI:10.1038/s41467-019-13755-5 |
| [29] |
Y. Xia, K.D. Gilroy, H.C. Peng, X. Xia, Angew. Chem. Int. Ed. 56 (2017) 60-95. DOI:10.1002/anie.201604731 |
| [30] |
G. Xiang, H. Guo, Y. Long, et al., Small 11 (2015) 4469-4474. DOI:10.1002/smll.201501088 |
| [31] |
H. Wang, H. Lin, Y. Long, et al., Nanoscale 9 (2017) 2074-2081. DOI:10.1039/C6NR09730H |
| [32] |
L. Liang, J. Yin, J. Bao, et al., Chin. Chem. Lett. 30 (2019) 167-170. DOI:10.1016/j.cclet.2018.01.049 |
| [33] |
H. Wang, H. Liu, Y. Ji, et al., Chem. Commun. 55 (2019) 2781-2784. DOI:10.1039/C8CC10093D |
| [34] |
Z. Li, Y. Wang, A.A. Elzatahry, et al., Chin. Chem. Lett. 31 (2020) 1598-1602. DOI:10.1016/j.cclet.2019.10.016 |
| [35] |
W.O, Y. Zhou, Y.J. Wang, H.Q. Wang, Z.B. Wu, Acta Phys. Chim. Sin. 37 (2021) 2009045. |
| [36] |
N. Wang, R.K. Miao, G. Lee, et al., SmartMat 2 (2021) 12-16. DOI:10.1002/smm2.1018 |
| [37] |
J. Zhang, H. Wang, L. Wang, et al., J. Am. Chem. Soc. 141 (2019) 2975-2983. DOI:10.1021/jacs.8b10864 |
| [38] |
Z. Zhang, H. Wang, M. Ma, et al., Chem. Eng. J. (2020) 127686. |
| [39] |
L. Jia, L.M. Yang, W. Wang, et al., Rare Met. (2019). DOI:10.1007/s12598-019-01241-2 |
| [40] |
J. Tian, Y. Sang, G. Yu, et al., Adv. Mater. 25 (2013) 5075-5080. DOI:10.1002/adma.201302014 |
| [41] |
C. Luo, X. Ren, Z. Dai, et al., ACS Appl. Mater. Inter. 9 (2017) 23265-23286. DOI:10.1021/acsami.7b00496 |
| [42] |
J.B. Li, X. Wu, S.W. Liu, Acta Phys. -Chim. Sin. 37 (2021) 200903038. |
| [43] |
D.O. Scanlon, C.W. Dunnill, J. Buckeridge, et al., Nat. Mater. 12 (2013) 798-801. DOI:10.1038/nmat3697 |
| [44] |
Z.C. Zhang, B. Xu, X. Wang, Chem. Soc. Rev. 43 (2014) 7870-7886. DOI:10.1039/C3CS60389J |
| [45] |
W.J. Yang, J.J. Li, X.Y. Cui, et al., Chin. Chem. Lett. 32 (2021) 2489-2494. DOI:10.1016/j.cclet.2020.12.057 |
| [46] |
H. Anwer, A. Mahmood, J. Lee, et al., Nano Res. 12 (2019) 955-972. DOI:10.1007/s12274-019-2287-0 |
| [47] |
J. Sun, M. Zhang, Z.F. Wang, et al., Rare Met. 38 (2019) 287-291. DOI:10.1007/s12598-014-0329-9 |
| [48] |
M. Wang, S. Zhang, Z.F. Du, et al., Rare Met. 38 (2019) 316-320. DOI:10.1007/s12598-014-0433-x |