 2021, Vol. 32
2021, Vol. 32 


b Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China;
c School of Materials and Chemical Engineering, Ningbo University of Technology, Ningbo 315211, China;
d Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China;
e CAS Center for Excellence in Nanoscience, Beijing Key Laboratory of Micro-Nano Energy and Sensor, Beijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences, Beijing 100083, China;
f School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453002, China
Halogen…halogen interactions in crystal engineering have received a massive number of attentions in the past several decades [1-3]. Though the halogen…halogen interactions belong to weak interactions, the intermolecular distances between halogens in crystals are significantly smaller than those of van der Waals interaction [4, 5]. With this view, halogen…halogen interactions play an important role in crystallization [6-8].
With the utilizing of scanning tunnelling microscope (STM), the halogen…halogen interactions in two-dimensional (2D) crystal engineering also have been wildly studied in recent years [9-15]. Currently, most of the recent studies about the halogen…halogen interactions at the liquid-solid interface are focused on the interactions between the same halogen atoms [16-18] and interactions between halogens and non-halogens [19, 20], while rare studies are related to the halogen…halogen interactions between different halogen atoms [21].
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) and its derivatives are widely applied to fluorescent sensors, organic solar cells, and other related areas [22-25] due to their excellent photoelectric performance and good chemical stability [22, 24, 25]. In a previous work, we used the BODIPY derivative, H-T-BO, as donor material and C70 molecule as acceptor material to construct a host-guest system at the 1-phenyloctane/highly oriented pyrolytic graphite (HOPG) interface and studied the structure on the molecular level by STM/STS method [26].
In this work, two aza-BODIPY derivatives, 3,5-di(p-bromophenyl-1,7-di(p-dodecyloxyphenyl)-aza-BODIPY (2Br-CJF) and 3,5-diphenyl-1,7-di(p-dodecyloxyphenyl)-aza-BODIPY (CJF), were synthesised (Scheme S1 in Supporting information) to investigate the role of halogen…halogen interactions in the formation of 2D molecular self-assembly architectures at 1-phenyloctane/HOPG interface. The chemical structure of aza-BODIPY is similar to that of BODIPY, but the 8-C atom is replaced by an N atom in aza-BODIPY [22]. In 2Br-CJF molecules, two 4-bromophenyls are modified on 3 and 5 position of the aza-BODIPY core, and two dodecyloxyphenyls on 1 and 7 position. The BF2 group of the aza-BODIPY and the bromine atom provide good sites to form halogen…halogen interactions. CJF molecule has a similar structure to 2Br-CJF molecule, but there is no bromine atom on the benzene ring. With this difference of molecular structure, we can compare the self-assembled patterns of these molecules and figure out how the Br…F-BF interaction influences the self-assembly. The self-assembly structures of 2Br-CJF and CJF on HOPG are investigated by STM. STM is a very important surface analysis technique because of its capability of capturing the position-specific image on the molecular level on surface [27-32]. The self-assembly behaviours and mechanisms are also investigated by density functional theory (DFT) calculations. The chemical structures of 2Br-CJF and CJF are shown in Scheme 1.

|
Download:
|
| Scheme 1. Chemical structures of 2Br-CJF and CJF. | |
The excremental methods are listed here. First, about the sample preparation, 1-phenyloctane was purchased from J&K Co., Ltd. All these materials were used without further purification. 1-Phenyloctane functioned as solvent to dissolve CJF and 2Br-CJF, and the concentrations of CJF and 2Br-CJF were controlled below 10−4 mol/L. The assemblies were prepared by subsequent deposition of the components onto a freshly cleaned HOPG (grade ZYB, SPI, U. S. A.) surface. CJF or 2Br-CJF solution was deposited on HOPG, and detected by STM. All experiments were performed at room temperature.
Next, is about the STM detection. The STM detection were performed on a Nanoscope IIIa (Bruker, U. S. A.) under ambient conditions. All the images were recorded using the constant-current mode at liquid/solid interface. The STM probes were made by mechanically cut Pt/Ir wires (80/20).
As for the DFT calculation. All theoretical calculations were performed using DFT-D scheme provided by DMol3 code. We used the periodic boundary conditions (PBC) to describe the 2D periodic structure on the graphite in this work. The Perdew-Burke-Ernzerhof parameterization of the local exchange correlation energy was applied in the generalized gradient approximation (GGA) to describe exchange and correlation [33]. All-electron spin-unrestricted Kohn-Sham wave functions were expanded in a local atomic orbital basis. For the large system, the numerical basis set was applied. All calculations were all-electron ones, and performed with the medium mesh. Self-consistent field procedure was done with a convergence criterion of 10−5 a.u. on the energy and electron density. Combined with the experimental data, we have optimized the geometry of the adsorbates in the unit cell. When the energy and density convergence criterion were reached, we could obtain the optimized parameters and the interaction energy between adsorbates. To evaluate the interaction between the adsorbates and HOPG, we design the model system. Since adsorption of benzene on graphite and graphene should be very similar [34], we had performed our calculations on infinite graphene monolayers using PBC. Considering that the interaction between adsorbates and substrate was mainly van der Waals interaction, the Grimme's dispersion corrections were adopted in our calculations. In the superlattice, slabs were separated by 35 Å in the normal direction. When modelling the adsorbates on graphene, we used graphene supercells and sampled the Brillouin zone by a 1 × 1 × 1 k-point mesh.
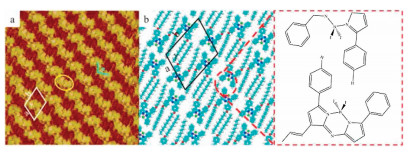
After depositing a droplet of CJF in 1-phenyloctane solution on the freshly cleaved HOPG, the self-assembly structure of the CJF molecule was formed at the 1-phenyloctane/HOPG interface. Then the large-scale domains were detected by STM (Fig. S16a in Supporting information). The CJF molecules formed a well-aligned and stable pattern on a large scale. Regular zig-zag lines were observed on the HOPG surface, and a high-resolution STM image is resolved for more details (Fig. 1a). Single CJF molecule presented as a bright H-like shaped spot. The CJF molecules alternated with each other in a dimer (marked by a yellow circle in Fig. 1a). As the phenyl groups are linked with the aza-BODIPY core by a single bond, the phenyl groups are not parallel to the HOPG strictly. So, the π-π interactions between the phenyl groups of CJF molecules in a dimer and the edge-to-face π-π interactions between the phenyl groups and HOPG are the driven forces to the formation of the dimers. Moreover, different dimers self-assembled into a zig-zag line. Obviously, the π-π interactions between the fastigiate phenyl groups from adjacent dimers facilitated the formation of the zig-zag lines. On another side of the CJF molecule, the darker strips staggered with each other in the zig-zag structure and the lengths were measured to be 1.45 ± 0.05 nm that were almost identical with those of dodecyl chains. The intermolecular van der Waals forces between long alkyl chains also helped to stabilize the zig-zag structure on the HOPG substrate. Fig. 1b is the corresponding molecular model given by DFT calculations based on the STM observations. A measured unit cell was marked in the STM image and the corresponding model. The measured parameters of unit cell were: a = 2.5 ± 0.1 nm, b = 3.1 ± 0.1 nm, γ = 108° ± 2° (Table 1).

|
Download:
|
| Fig. 1. (a) High-resolution STM image of CJF self-assembled structures at PO/HOPG interface (Iset = 271.6 pA, Vbias = 816.3 mV, 20.4 nm × 20.4 nm). (b) Proposed molecular model of CJF. Inset illuminates that there is no interaction in H atoms and F atoms. | |
|
|
Table 1 The experimental (exptl.) and calculated (calcd.) unit cell parameters for the CJF cells and 2Br-CJF cells. |

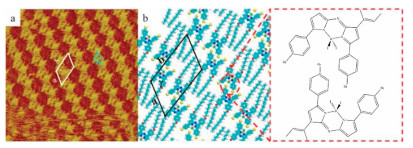
To further explore if Br…F-BF interaction could influence the self-assembly of CJF molecules, we observed the self-assembly structure of 2Br-CJF molecules with STM. The same with CJF molecules, a droplet of the 2Br-CJF solution in 1-phenyloctane was deposited on the freshly cleaved HOPG. Then the large-scale structure was detected by STM (Fig. S16b in Supporting information). Due to the similar molecular structure with CJF, the 2Br-CJF molecules formed a similar self-assembly pattern with CJF molecules. Each H-like bright spot corresponded to a 2Br-CJF molecule, and dimers composed of two 2Br-CJF molecules self-assembled into regular zig-zag lines on the HOPG surface. However, after detecting the high-resolution STM image (Fig. 2a), we can clearly distinguish some different details of 2Br-CJF's self-assembly structure from CJF's. First of all, the measured cell parameters were: a = 2.3 ± 0.1 nm, b = 3.6 ± 0.1 nm, γ = 119° ± 2° (Table 1). The differences in cell parameters indicated that the driven force may be different in the 2Br-CJF self-assembly structure. Except for the π-π interactions we mentioned above, the halogen…halogen interactions between Br atoms and F atoms of BF2 groups also affected 2Br-CJF's self-assembly structure. A pair of Br…F-BF interactions exist between the Br atoms of 4-bromophenyls and the F atoms of BF2 groups on two adjacent molecules. The BF2 group is an electron-deficient group, and the Br atom has a lone electron pair. So, in two 2Br-CJF molecules, there are two antiparallel Br…F-BF interactions. One is formed by one F atom of the BF2 group on the first 2Br-CJF molecule and one Br atom on the second 2Br-CJF molecule. Another one is formed by the Br atom of the first 2Br-CJF molecule and one F atom of the BF2 group on the second2Br-CJF molecule. Next, we measured the angle in the zig-zag lines (α, Marked with blue lines in Figs. 1a and 2a). In the CJF self-assembly structures, the angle α was measured to be 114° ± 2°. While in the self-assembly structure of 2Br-CJF, the angle α was measured to be 104° ± 2°. The smaller angle of the zig-zag lines indicated that the phenyl group of a 2Br-CJF molecule was closer to the axis of symmetry of another 2Br-CJF molecule. This further demonstrated that the Br…F-BF interaction has changed the self-assembly structure of the aza-BODIPY molecules.

|
Download:
|
| Fig. 2. (a) High-resolution STM image of 2Br-CJF (Iset = 299.1 pA, Vbias = 770.3 mV, 21.9 nm × 21.9 nm). (b) Proposed molecular model of 2Br-CJF. Inset illuminates the Br…F—BF interaction as indicated by dotted lines. | |
In order to further demonstrate the result, DFT calculations for the CJF's and the 2Br-CJF's assembly system were conducted based on the observed phenomena. The calculated parameters are listed in Table 1. The calculated parameter agreed with the experimental data. In the surface-assisted self-assembly system, the interaction between adsorbates and the interaction between adsorbates and substrates are important. Therefore, we presented the total energy including the interaction energy between adsorbates and the interaction energy between adsorbates and the substrate. These results are the total energy of a cell. We could compare the total energy to evaluate the thermodynamic stability of different systems in general. However, if we need to compare two systems with different unit cells precisely, we should consider the effect of the unit area. So, a more reasonable way to compare the thermodynamic stability of the different systems should be the total energy per unit area. All of the calculation results we mentioned above are listed in Table 2.
|
|
Table 2 Total energies and energies per unit area of the assembled structures of CJF and 2Br-CJF. |

{kind=link}
{kind=link}
{kind=link}
From Table 2, we notice that the interaction energy between adsorbates of 2Br-CJF assembled structure is −43.172 kcal/mol, which is lower than that between adsorbates of CJF assembled structure (−28.173 kcal/mol). It means that the interaction between the 2Br-CJF molecules is stronger, which strongly supports the existence of Br…F interaction. Combining with the differences in the cell parameter of the 2Br-CJF and CJF assembled system we presented above, we suggest the Br…F-BF interaction between the 2Br-CJF dimers plays an important role in the self-assembly process. The Br…F-BF interaction strengthens the total interaction between 2Br-CJF dimers and changes the 2Br-CJF self-assembled structure. There is no significant difference in the interactions between adsorbates and substrate, which is acceptable because the molecular structures of these two molecules are similar. The tiny difference in the interaction between adsorbates and substrate may be caused by the Br-π interactions of 2Br-CJF. This kind of halogen…π interaction has ever been reported in many previous works [35-37]. The total energy per unit area of the 2Br-CJF assembled system is −0.229 kcal mol−1 Å −2, lower than that of the CJF assembled system (−0.205 kcal mol−1 Å −2). It also means that the 2Br-CJF assembled system is an energetically favourable structure. In this system, the Br…F-BF interaction facilitates the stability of the self-assembled structure. Moreover, in the optimized molecular model of CJF system and 2Br-CJF system (Figs. 1b and 2 b, respectively), we can notice that the distance between the F atoms and H atoms of CJF (marked with a red circle in Fig. 1b) is 0.51 ± 0.01 nm, which is larger than the van der Waals radii of these two atoms. So, we think there is no interaction between the F and H atoms. The distance between the F atoms and Br atoms of 2Br-CJF (Marked with red dotted lines in the inset of Fig. 2b) is 0.30 ± 0.01 nm, this length agreed to the length of halogen…halogen interactions reported in the previous article [20], indicating that there are halogen…halogen interactions between the Br atoms and F atoms of BF2 groups.
In addition, the energy analysis based on the whole unit cell provides limited information about the Br…F-BF halogen interaction, as it may be overwhelmed by the contribution from the difference at the alkyl chains, aromatic part. In order to exclude the possibility that the structure is only driven by close-packing and the interactions between alkyl chains and aromatics, based on the previous work we build a high-quality DFT calculation model with only key atoms (Fig. S17 in Supporting information) for such interaction motif [38]. In this DFT calculation, we calculated the energy of the Br…F-BF interaction is −1.256 kcal/mol. This means the Br…F-BF interaction is a week interaction in this self-assembly system.
In conclusion, we investigated self-assembled structures of two aza-BODIPY derivatives, CJF molecule and 2Br-CJF molecule with STM and demonstrated that the Br…F interactions exist and can adjust the 2D self-assembly behaviour of the aza-BODIPY derivatives at the 1-phenyloctane/HOPG interface. This result was supported by the DFT calculation. We expect that this research can enrich the method of designing the 2D molecular networks and provide a new idea to explain some self-assembly behaviours of halides on the solid-liquid interface.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Basic Research Program of China (No. 2016YFA0200700), the National Natural Science Foundation of China (Nos. 21773041, 21972031), the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB36000000), the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University, National Science Foundation of Zhejiang Province of China (No. Y20B020032) and Project of Ningbo Science and Technology Innovation 2025.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j. cclet.2021.02.066.
| [1] |
K. Reichenbacher, H.I. Suss, J. Hulliger, Chem. Soc. Rev. 34 (2005) 22-30. DOI:10.1039/B406892K |
| [2] |
T.T. Bui, S. Dahaoui, C. Lecomte, G.R. Desiraju, E. Espinosa, Angew. Chem. Int. Ed. 48 (2009) 3838-3841. DOI:10.1002/anie.200805739 |
| [3] |
S.K. Nayak, M.K. Reddy, T.N.G. Row, D. Chopra, Cryst. Growth Des. 11 (2011) 1578-1596. DOI:10.1021/cg101544z |
| [4] |
G.R. Desiraju, R. Parthasarathy, J. Am. Chem. Soc. 111 (1989) 8725-8726. DOI:10.1021/ja00205a027 |
| [5] |
V.R. Pedireddi, D.S. Reddy, B.S. Goud, et al., J. Chem. Soc. Perkin Trans. 2 (1994) 2353-2360. DOI:10.1039/P29940002353 |
| [6] |
E. Albright, J. Cann, A. Decken, S. Eisler, CrystEngComm 19 (2017) 1024-1027. DOI:10.1039/C6CE02339H |
| [7] |
J.S. Ovens, D.B. Leznoff, CrystEngComm 20 (2018) 1769-1773. DOI:10.1039/C7CE02167D |
| [8] |
S.A. Adonin, M.A. Bondarenko, A.S. Novikov, et al., CrystEngComm 21 (2019) 6666-6670. DOI:10.1039/C9CE01106D |
| [9] |
F. Silly, J. Phys. Chem. C 117 (2013) 20244-20249. DOI:10.1021/jp4057626 |
| [10] |
Z. Guo, P. Yu, K. Sun, et al., Phys. Chem. Chem. Phys. 19 (2017) 31540-31544. DOI:10.1039/C7CP06027K |
| [11] |
B. Zha, J. Li, J. Wu, X. Miao, M. Zhang, New J. Chem. 43 (2019) 17182-17187. DOI:10.1039/C9NJ04726C |
| [12] |
J. Hou, B. Tu, Q. Zeng, C. Zhan, J. Yao, Chin. Chem. Lett. 31 (2020) 353-356. DOI:10.1016/j.cclet.2019.07.029 |
| [13] |
X. Gong, J. Zhang, S. Jiang, Chem. Comm. 56 (2020) 3054-3057. DOI:10.1039/C9CC08768K |
| [14] |
F. Guan, Z. Song, F. Xin, et al., J. Bioresour. Bioprod. 5 (2020) 37-43. DOI:10.1016/j.jobab.2020.03.004 |
| [15] |
D.W. Wei, H. Wei, A.C. Gauthier, et al., J. Bioresour. Bioprod. 5 (2020) 1-15. DOI:10.1016/j.jobab.2020.03.001 |
| [16] |
A. Mukherjee, A. Sanz-Matias, G. Velpula, et al., Chem. Sci. 10 (2019) 3881-3891. DOI:10.1039/C8SC04499F |
| [17] |
B. Zha, M. Dong, X. Miao, et al., Nanoscale 9 (2017) 237-250. DOI:10.1039/C6NR07693A |
| [18] |
B. Zha, X. Miao, P. Liu, Y. Wu, W. Deng, Chem. Commun. 50 (2014) 9003-9006. DOI:10.1039/C4CC03687E |
| [19] |
Q.N. Zheng, X.H. Liu, T. Chen, et al., J. Am. Chem. Soc. 137 (2015) 6128-6131. DOI:10.1021/jacs.5b02206 |
| [20] |
R. Gutzler, O. Ivasenko, C. Fu, et al., Chem. Commun. 47 (2011) 9453-9455. DOI:10.1039/c1cc13114a |
| [21] |
M. Dong, K. Miao, J. Wu, et al., J. Phys. Chem. C 123 (2019) 4349-4359. DOI:10.1021/acs.jpcc.8b12176 |
| [22] |
A. Loudet, K. Burgess, Chem. Rev. 107 (2007) 4891-4932. DOI:10.1021/cr078381n |
| [23] |
N. Boens, V. Leen, W. Dehaen, Chem. Soc. Rev. 41 (2012) 1130-1172. DOI:10.1039/C1CS15132K |
| [24] |
T. Bura, N. Leclerc, S. Fall, et al., J. Am. Chem. Soc. 134 (2012) 17404-17407. DOI:10.1021/ja3072513 |
| [25] |
W. Liu, A. Tang, J. Chen, et al., ACS Appl. Mater. Interfaces 6 (2014) 22496-22505. DOI:10.1021/am506585u |
| [26] |
J. Xu, W. Liu, Y. Geng, et al., Nanoscale 9 (2017) 2579-2584. DOI:10.1039/C6NR08234C |
| [27] |
J. Xu, Y. Li, L. Wang, et al., Chin. Chem. Lett. 30 (2019) 767-770. DOI:10.1016/j.cclet.2018.09.019 |
| [28] |
J. Li, X. Zu, Y. Qian, et al., Chin. Chem. Lett. 31 (2020) 10-18. DOI:10.1016/j.cclet.2019.04.032 |
| [29] |
J. Li, Y. Qian, W. Duan, Q. Zeng, Chin. Chem. Lett. 30 (2019) 292-298. DOI:10.1016/j.cclet.2018.05.037 |
| [30] |
Q. Xue, Y. Zhang, R. Li, et al., Chin. Chem. Lett. 30 (2019) 2355-2358. DOI:10.1016/j.cclet.2019.09.027 |
| [31] |
J. Hou, H. Dai, Z. Zhang, et al., Langmuir 33 (2017) 400-406. DOI:10.1021/acs.langmuir.6b03567 |
| [32] |
X. Zhu, Y. Geng, H. Xiao, et al., Langmuir 34 (2018) 5169-5173. DOI:10.1021/acs.langmuir.7b04293 |
| [33] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [34] |
J.M. MacLeod, J.A. Lipton-Duffin, D. Cui, S. De Feyter, F. Rosei, Langmuir 31 (2015) 7016-7024. DOI:10.1021/la5048886 |
| [35] |
H. Sun, A. Horatscheck, V. Martos, et al., Angew. Chem. Int. Ed. 56 (2017) 6454-6458. DOI:10.1002/anie.201700520 |
| [36] |
H.G. Wallnoefer, T. Fox, K.R. Liedl, C.S. Tautermann, Phys. Chem. Chem. Phys. 12 (2010) 14941-14949. DOI:10.1039/c0cp00607f |
| [37] |
I.S. Youn, D.Y. Kim, J.W. Cho, et al., J. Phys. Chem. A 120 (2016) 9305-9314. DOI:10.1021/acs.jpca.6b09395 |
| [38] |
L.N. Anderson, F.W. Aquino, A.E. Raeber, X. Chen, B.M. Wong, J. Chem. Theory Comput. 14 (2018) 180-190. DOI:10.1021/acs.jctc.7b01078 |