 2021, Vol. 32
2021, Vol. 32 


b College of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
Semiconductor-based photocatalysis for hydrogen production, which provides clean hydrogen energy, is considered to be a sustainable solution for the alleviation of energy and environmental issues [1]. The efficient capture of solar energy is the first step in obtaining an ideal photocatalyst. Near-infrared (NIR) light accounts for more than half of the incoming solar flux (~55%). Nevertheless, it has a low photon energy, and suitable redox potentials required for photoinduced redox reactions are still difficult to be achieved. Therefore, the effective utilization of NIR light for photocatalytic reactions, especially for the H2 generation via water splitting remains a challenge. Since 2009, polymeric carbon nitride (PCN) has been considered as a metal-free model candidate for solar energy conversion, mostly because of the controllable bandgap, suitable band position, and tailorable texture [2-12]. However, the light response of bulk PCN is limited to the visible region (~450 nm), resulting in an insufficient photocatalytic activity.
Three main strategies have been developed to improve the NIR photon capture: engineering of an optical gap [13-16], decoration with noble metals or upconversion material [17-19] and element doping via small molecular copolymerization with melamine [20-23]. Among the strategies, the incorporation of nonmetallic heteroatoms (C, O, P, S, B and F) into the PCN framework effectively regulates the optical gap and improves the photocatalytic performance [24-26]. Usually, an interband level generally forms between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LOMO), which results in the capture of NIR light [14]. In addition, C-doping of PCN has great potential for the amelioration of the photoactivity and broadening of the light absorption. Experimental and theoretical calculations showed that the substitution of N in the heptazine ring with C atoms leads to the formation of a delocalized π-conjugation, thereby enhancing the electronic conductivity and photocatalytic performance [24, 27]. In most cases, the light absorption was extended to the NIR region; however, the photocatalytic activity did not improve under irradiation with NIR light. This has been mainly explained with the surface modification and heterogeneous distribution of the dopants, which can only act as structural defects and/or recombination centers of photoexcited charge carriers. However, it remains unclear whether the photocatalytic activity in the extended light range can be further improved; there is a lack of sufficient studies regarding this topic. In a study, bulk doping of the host lattice was carried out based on which the bandgap could be narrowed and the light range was broadened [28]. Recent reports showed that PCN synthesized via alkali-assisted route (e.g., NaBH4 [29], KOH [30], NaOH [31], K2CO3 [32] and Na2CO3 [33]) exhibits an enhanced visible-light photocatalytic performance for water splitting. The enhanced activity is due to the introduction of surface defects and changes of the HOMO or LOMO positions, the improved dispersion of PCN in H2O and broaden the light capture range [34]. Considering the hydrophobicity of carbon-doped PCN (cPCN) obtained by direct polycondensation at high temperature and the "molecular scissors" function of alkalis, we combined carbon doping (1, 3, 5-trimesic acid, TA, as the dopant) with alkali activation to bifunctionalize PCN for an enhanced photocatalytic activity under irradiation with NIR light (λ ≥ 700 nm) for H2 evolution and H2O2 production from H2O.
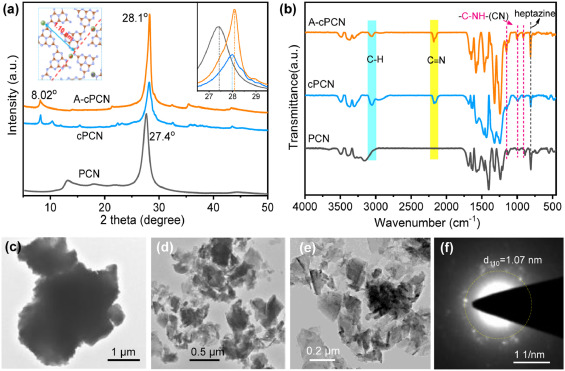
The process for the preparation of cPCN and A-cPCN was shown in Fig. S1 (Supporting information). The change in the structure of cPCN before and after activation by K2CO3 was analyzed with X-ray diffraction (XRD), as shown in Fig. 1a. The diffraction peak at 8.02°, which is associated with the (110) facet of A-cPCN and referring to the distance between K+ cations inserted into PCN melon chains [35] (inset in left in Fig. 1a), was maintained after K2CO3 activation while the peak at 13° was disappeared that in PCN. In addition, the main diffraction peak centered at 28.1°, which corresponds to the intrinsic layered structure of carbon nitride sharply increases, indicating the significantly improved crystallinity of A-cPCN. The increased crystalline means more regular alignment and a long-range order of melon chains in A-cPCN, thus leading to a decreased layer distance than the amorphous PCN. The full width at half maximum (FWHM) of the (002) diffraction peak (28.1°) is 0.66 in A-cPCN, whereas that is 1.48 in cPCN and 1.56 in PCN, further indicating the improved crystallinity. Fig. S2 (Supporting information) shows that the use of different TA concentrations for the synthesis of cPCN and then activated by K2CO3 that leads to an increased crystallinity. Direct treatment of pristine PCN with K2CO3 does not yield an improved crystallinity (Fig. S3 in Supporting information). The Fourier transform infrared (FTIR) spectra was used to inquiry the changes after alkali activation (Fig. 1b and Fig. S4 in Supporting information). New peaks located at 2182 and 2157 cm−1 in cPCN and A-cPCN can be assigned to the stretching vibration of cyano units (C≡N), probably due to the incomplete polymerization or decomposition of heptazines [35]. Transmission electron microscopy (TEM) shows that cPCN consists of large particles with sizes at the micrometer scale (Fig. 1c) and an amorphous nature as demonstrated by the selected area electron diffraction (SAED) pattern (Fig. S5 in Supporting information). In contrast, after activation with K2CO3, the large bulk particles become smaller (100–400 nm) and display a nanosheet texture (Figs. 1d and e). The SAED patterns of A-cPCN display conspicuous bright spots (Fig. 1f). One set of spots marked by a yellow ring can be indexed to the crystalline facet of (110) with a spacing of 1.07 nm. This corresponds to the XRD peak at 8°, indicating that the crystallinity of A-cPCN improved after the activation with K2CO3.

|
Download:
|
| Fig. 1. (a) XRD patterns and (b) FT-IR spectra of PCN, cPCN, and A-cPCN. TEM and images of cPCN (c) and A-cPCN (d–e). SAED patterns of A-cPCN (f). | |
{kind=link}
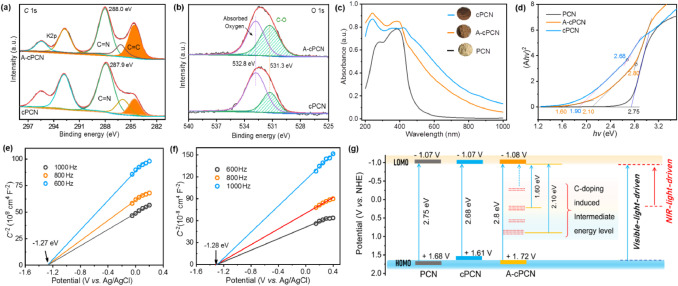
X-ray photoelectron spectroscopy (XPS) survey view show that C, N, O and K are present in both cPCN and A-cPCN (Figs. S6 and S7 in Supporting information). The sp2 hybridized aromatic C atoms (C=N) in the heptazine units in cPCN and A-cPCN insignificantly change based on the C 1s XPS spectra (Fig. 2a). The peak centered at 284.6 eV, which corresponds to the C=C/C–C components, significantly increases in A-cPCN (34%) and cPCN (17%) compared with that in pure PCN (9.4%; Table S1 in Supporting information). This validates that the C atom was successfully introduced into the crystal structure. In addition, the percentage of the C=C peak in A-cPCN is double higher than that of cPCN, which can be ascribed to the purification of cPCN by K2CO3. During this process, small molecular oligomers or fragments of cPCN are degraded into gas or soluble ions. The positively charged K ions can also break the C and N bonds in heptazine rings [34], leading to the formation of new structural edges with functional groups such as C≡N, –NH2, and C–OH (Fig. S4) [36]. In the high-resolution N 1s spectra in (Fig. S6b in Supporting information), the feature at ~398.6 eV, corresponding to –NHx at the edges of heptazine substructures, increases in A-cPCN compared with cPCN. The high-resolution O 1s signals (Fig. 2b) further indicate that both A-cPCN and cPCN contain oxygen (C−OH, 531.3 eV). The intensity of the signal obtained for A-cPCN is much higher than cPCN, which would benefit for the better dispersibility of A-cPCN in water [37].

|
Download:
|
| Fig. 2. High-resolution XPS spectra C 1s (a), and O 1s (b) of cPCN and A-cPCN. UV–vis–NIR absorption spectra (c) and Kubelka-Munk plots (d) of PCN, cPCN, and A-cPCN. Mott–Schottky plots of cPCN (e) and A-cPCN (f). Schematic of the energy level structure for PCN, cPCN, and A-cPCN (g). | |
{kind=link}
Carbon doping combined with alkali activation obviously changed the optical/electronic properties of PCN, as revealed by the UV–vis–NIR absorption spectra (Fig. 2c). C-doped carbon nitrides (cPCN and A-cPCN) show a prominent redshift from 400 nm to 1000 nm in comparison with pristine PCN, which is in line with the color alteration of cPCN (reddish brown) and A-cPCN (light brown; insets in Fig. 2c). This might be due to the incorporation of the C=C chromophores in cPCN and A-cPCN, which broadens the π-conjugated systems and introduces new intermediate bands, thereby narrowing the bandgap. However, the light absorption edge exhibits a blueshift after K2CO3 treatment, which may be ascribed to the decrease in the size of the nanosheets. Moreover, the extended light absorption with a flat tail in cPCN and A-cPCN is not caused by the narrowed bandgap but by the inhomogeneous doping of carbon, which causes localized states in the bandgap (interband energy levels), enabling the capture of NIR light [25-27].
The Kubelka-Munk plots in Fig. 2d reveal two more bandgap energies. The bandgap of PCN is approximately 2.75 eV, whereas the bandgap energies of cPCN are 2.68 and 1.90 eV and those of A-cPCN are 2.80, 2.10, and 1.60 eV. The new energy levels (2.1 and 1.6 eV) between the HOMO and LOMO in A-cPCN are due to the carbon doping. This is the key to the expansion of the light absorption range. The flat-band potentials of cPCN and A-cPCN were also analyzed using Mott–Schottky plots (Figs. 2e and f). Ideally, the conduction band potential (or LOMO position) of a semiconductor is approximately equal to the flat-band potential. Therefore, the LOMO positions of cPCN and A-cPCN are −1.07 and −1.08 eV, respectively. Based on those results, as illustrated in Fig. 2g, the LOMO positions of cPCN and A-cPCN are almost the same as that of PCN, which indicates that the reduction potential insignificantly changed after K2CO3 activation. For A-cPCN and cPCN, the electrons in the intermediate energy levels induced by the doped carbon can be excited under visible and NIR light irradiation.
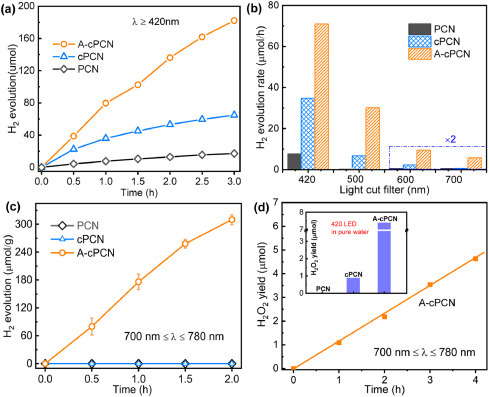
The activity of the photocatalysts was first evaluated by releasing H2 from water under vis–NIR light. Upon irradiation with visible light (780 nm ≥ λ ≥ 420 nm), A-cPCN exhibits the highest photocatalytic activity for H2 evolution (~3.9 mmol h−1 g−1) from water (Fig. 3a), which is approximately 2-fold that of cPCN and ten times higher than that of pure PCN (~0.4 mmol h−1 g−1). This reveals that C-doping effectively increases the ability for light capture, thereby greatly enhancing the activity for H2 production. Compared with cPCN, the photocatalytic activity doubles over A-cPCN, which can be ascribe to the solid alkali treatment decreases the particle size and correspondingly reduces the diffusion distance of photoexcited charge carriers from the bulk to the surface. Furthermore, the photocatalytic activity was determined under irradiation with NIR light. As shown in Fig. 3b and Fig. S8 (Supporting information), H2 evolution from water cannot be observed in pure PCN irradiated by blue light (780 nm ≥ λ ≥ 500 nm) and orange light (780 nm ≥ λ ≥ 600 nm) due to the limited light response. In contrast, the rate of H2 generation in A-cPCN is approximately 30.1 and 5.6 µmol/h under irradiation with blue and orange light, respectively, which is around five times that of cPCN. Furthermore, under illumination with NIR light (780 nm ≥ λ ≥ 700 nm), only A-cPCN exhibits a photocatalytic activity (Fig. 3c), and the gas evolution rate is about 3.3 µmol/h (20 mg catalyst). Additionally, a comparation study of photocatalytic hydrogen generation activity over cPCN with different content of carbon doping (controlled by the loading of TA precursor, Fig. S9 in Supporting information), as well as that before and after K2CO3 activation (Fig. S10 in Supporting information) evidently demonstrated that the indispensable role of K2CO3 in the promotion of photocatalytic performance. However, the alkali K2CO3 is a double-edged sword in modification of PCN. With low loading, K2CO3 would work as scissors to cut the big aggregates bulk carbon nitride into nanosheets. But high loading of K2CO3 would break down the chemical bands and even frameworks of PCN, as demonstrated by the widen diffraction peak at 28.1° in XRD spectra in A-cPCN3-5 (Fig. S2). The optimized loading is A-cPCN2. Furthermore, the chemical structure of A-cPCN2 is much stable after photocatalytic utilization, as demonstrated by XRD (Fig. S11 in Supporting information) and FT-IR (Fig. S12 in Supporting information).

|
Download:
|
| Fig. 3. (a) Photocatalytic H2 evolution over carbon nitrides under irradiation with visible light with a 420-nm light filter (780 nm ≥ λ ≥ 420 nm). (b) Comparison with the hydrogen production rate based on the use of different light filters and (c) photocatalytic H2 evolution under irradiation and (d) photocatalytic H2O2 production over carbon nitrides under irradiation with NIR light (780 nm ≥ λ ≥ 700 nm) with isopropanol as electron donor using 10 mg catalyst. The inset shows the result obtained under visible light irradiation (20 W, 420 nm LED) in pure water within the first hour. | |
{kind=link}
The reduction potentials (LOMO position, Fig. 2g) of the synthesized carbon nitrides are also enough for the reduction of O2 via two-electron route (O2 + e− → O2•− and O2•− + 2H+ + e− → H2O2, ψ(O2/H2O2) = 0.68 V vs. NHE) for the production of H2O2 [38]. The photocatalytic H2O2 generation was evaluated under monochromatic LED light (Fig. S13 in Supporting information). The inset in Fig. 3d shows that, without the cocatalyst and electron donor, the H2O2 yield in A-cPCN is 7.4 µmol after 1 h under irradiation with 420 nm LED light, which is 8 times that of cPCN. The yield obtained for pristine PCN is negligible. Furthermore, the H2O2 generation over A-cPCN at NIR light (780 nm ≥ λ ≥ 700 nm) in the 10% isopropanol system is about 1.09 µmol at the first 1 h irradiation. No H2O2 signal was observed even for concessive 3 h irradiation under NIR light over PCN. These results indicate that alkali activation of carbon-doped carbon nitride plays a vital and synergistic role in narrowing the band gap, broadening the light absorption, and achieving an enhanced photocatalytic performance under NIR light.
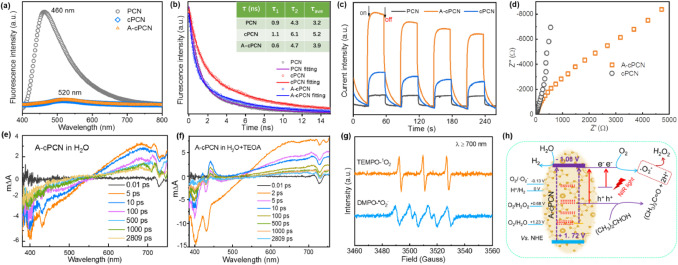
Fig. S14a (Supporting information) shows that the contact angles of pristine PCN, cPCN and A-cPCN are approximately 33°, 13°, and 14°, respectively, indicating the enhanced hydrophilicity of cPCN and A-cPCN. Theoretically, both A-cPCN and cPCN should homogenously disperse in water, which can be clearly observed in Fig. S14b (Supporting information), whereas cPCN sediments can be found at the bottom of the beaker, which is mainly due to the large particle size. Additionally, the steady-state photoluminescence (PL) spectroscopy (Fig. 4a) shows a redshift of the main PL emission peak of c-PCN and A-cPCN than PCN. This is consistent with the UV-vis-NIR absorption results and can be ascribed to the incorporation of C=C into the heptazine units, resulting in the expansion of the π-electron conjugated system. Furthermore, compared with PCN, both cPCN and A-cPCN display significantly decreased PL intensities, suggesting the effective separation of photoexcited charge carriers, which leads to a higher photocatalytic activity (Fig. 3). Time-resolved transient fluorescence decay spectra were recorded to further reveal the migration efficiency of photogenerated charge carriers. The lifetimes of A-cPCN (3.9 ns) and cPCN (5.2 ns) are longer than that of PCN (3.2 ns) (Fig. 4b), which indicates that much more efficient electron-hole separation, thus why both of A-cPCN and cPCN show a much higher photocatalytic activity for hydrogen evolution. However, the PL lifetimes of A-cPCN (3.9 ns) is much shorter than that of cPCN (5.2 ns). By considering near 5-fold enhanced photocatalytic activity of A-cPCN than cPCN, we ascribed that the shorter PL lifetime is due to the accelerated exciton dissociation at the abundant order-disorder interfaces in crystalline A-cPCN, which is good agreement with the finding by Xie group [39].

|
Download:
|
| Fig. 4. Steady-state photoluminescence spectra (a), time-resolved fluorescence decay spectra (b) and photocurrent intensity curves (c) of PCN, cPCN, and A-cPCN. (d) EIS Nyquist plots of cPCN and A-cPCN. 365 nm nanosecond laser was used as the excited source in Figs. 4a and b. Transient absorption spectra obtained from suspensions of A-cPCN in pure H2O (e) and a solvent mixture of 10 vol% of TEOA in H2O (f). All data were obtained using an excitation wavelength of 365 nm. (g) EPR spectra of the DMPO-O2•− and TEMP-1O2 adducts of A-cPCN under irradiation with NIR light (λ ≥ 700 nm). (h) Proposed reaction mechanism based on energy-band engineering for the photoproduction of H2 and H2O2 over A-cPCN under NIR light. | |
{kind=link}
The results of surface photocurrent responses (Fig. 4c) of cPCN and A-cPCN further confirms the efficient separation and rapid migration of charge carriers to the surface. The electrochemical impedance spectroscopy (EIS) shows that the arc radius of A-cPCN is much smaller than that of cPCN (Fig. 4d), suggesting that the incorporation of C=C building blocks in the carbon nitride framework decreases the electron transport resistance, thereby leading to a high photocatalytic activity, similar to P-doped PCN [40].
Finally, the dynamics of the excited states of carbon nitrides and the role of TEOA in the photocatalytic water splitting process was investigated by employing the transient absorption spectroscopy (TAS). Figs. 4e, f and Fig. S15 (Supporting information) show transient spectra obtained 2 ps to 2.8 ns after the excitation (365 nm) of A-cPCN suspensions in pure H2O (Fig. 4e) and the reaction mixture H2O/TEOA (Fig. 4f). Immediately after the excitation, the transient spectra exhibit a bleaching signal (negative signal) between 380 and 550 nm and excited-state absorption signal above 650 nm (positive absorption). In the H2O/TEOA system, the TAS spectra evolve within the first ~5 ps; a new prominent positive absorption feature appears at ~420 nm with a half-time of 574 ps (Fig. S16 in Supporting information). This feature might be due to an electron polaron, which is the active species for hydrogen production in A-cPCN in the presence of a hole scavenger (TEOA). In the reaction mixture and H2O, another feature appears at ~650 nm within pico- to nanoseconds. This feature may be related to an exciton with increased charge transfer (photogenerated electron) in A-cPCN. These observations are in good agreement with reported polaron signatures of polymers in pure H2O and scavengers [41].
In addition, the generation of reactive species in the NIR-active A-cPCN was analyzed using the spin-trapping electron paramagnetic resonance technique with capturing agents of 2, 2, 6, 6-tetramethylpiperidine (TEMP) and 5, 5-dimethyl-1-pyrroline N-oxide (DMPO). Fig. 4g shows that the EPR signals were obtained under irradiation with NIR light (λ ≥ 700 nm) for 10 min. Two conspicuous ESR signals were obtained, with intensity ratios of 1:1:1 for the TEMP-1O2 adduct and 1:2:2:1 for the DMPO-O2•− adduct. The produced reactive species provide a route for H2O2 generation by photoreduction of O2, as the proposed mechanism shown in Fig. 4h. The charge transfer pathways during the photoproduction of H2 and H2O2 are mainly based on the inter-band formed by the doped carbon. Under NIR light irradiation, the electrons can not be excited from HOMO to LUMO in A-cPCN. However, multi-steps excitation and migration of electrons may be realized under NIR light over carbon doped A-cPCN. For example, the photogenerated electrons in HOMO may firstly transfer to the doped energy levels (Fig. 2g) and then to the LUMO of A-cPCN, or the electrons in the doped energy levels (1.6 eV) directly transfer to the LUMO of A-cPCN, and finally to reduction of water for H2 evolution. In the formation of H2O2, the produced hot electrons (−1.08 V vs. NHE) can only be excited from the localized electrons on the doped elements and followed by the reduction of the adsorbed molecular oxygen to generate O2•− species (O2/O2•−, −0.13 V vs. NHE).
In conclusion, a NIR-light-active A-cPCN photocatalyst was successfully synthesized for efficient H2 and H2O2 production from water under NIR light. The synthesized A-cPCN exhibits a significantly enhanced efficiency with respect to the separation of photoexcited charge carriers, enhanced dissociation of photogenerated excitons and decreased electrochemical impedance compared with cPCN and PCN. The carbon doped PCN framework leads to the generation of intermediate energy level and based on which photon energies ranging from the UV to the NIR region can be efficiently captured. Thus, in contrast to cPCN and PCN, A-cPCN exhibits an enhanced H2 evolution and H2O2 production from water under irradiation with NIR light (780 nm ≥ λ ≥ 700 nm). Although the photocatalytic efficiency is still low under NIR light, the combination of carbon doping with alkali activation will boost the development of low-cost photocatalysts for the efficient utilization of solar energy for artificial photocatalysis.
Declaration of competing interestThe authors declare that they have no known competing financial interests that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work has been supported by the National Natural Science Foundation of China (Nos. 21972094, 21902105, 21805191, 51701127, 21401190), Guangdong Special Support Program, Pengcheng Scholar Program, Shenzhen Innovation Program (Nos. JCYJ20170818142642395, JCYJ20190808142001745), and the Natural Science Foundation of Guangdong Province (No. 2020A1515010982).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.cclet.2021.05.039.
| [1] |
J.H. Kim, D. Hansora, P. Sharma, et al., Chem. Soc. Rev. 48 (2019) 1908-1971. DOI:10.1039/c8cs00699g |
| [2] |
W.J. Ong, L.L. Tan, Y.H. Ng, et al., Chem. Rev. 116 (2016) 7159-7329. DOI:10.1021/acs.chemrev.6b00075 |
| [3] |
C.B. Wu, G.H. Yu, Y. Yin, et al., Small 16 (2020) 2003162. DOI:10.1002/smll.202003162 |
| [4] |
K.Z. Qi, S. Liu, A. Zadad, J. Taiwan Inst. Chem. Eng. 109 (2020) 111-123. DOI:10.1016/j.jtice.2020.02.012 |
| [5] |
D.Y. Ni, Y.Y. Zhang, Y.F. Shen, et al., Chin. Chem. Lett. 31 (2020) 115-118. DOI:10.1016/j.cclet.2019.04.068 |
| [6] |
Y.S. Xu, W.D. Zhang, ChemCatChem 5 (2013) 2343-2351. DOI:10.1002/cctc.201300144 |
| [7] |
L.H. Lin, Z.Y. Lin, J. Zhang, et al., Nat. Catal. 3 (2020) 649-655. DOI:10.1038/s41929-020-0476-3 |
| [8] |
K. Qi, W. Lv, I. Khan, S. Liu, Chin. J. Catal. 41 (2020) 114-121. DOI:10.1016/S1872-2067(19)63459-5 |
| [9] |
P. Qiu, Z. Liang, X. Liu, et al., J. Coll. Interf. Sci. 571 (2020) 318-325. DOI:10.1016/j.jcis.2020.03.062 |
| [10] |
Y.S. Xu, C.T. Qiu, X. Fan, et al., Appl. Catal. B: Environ. 268 (2020) 118457. DOI:10.1016/j.apcatb.2019.118457 |
| [11] |
F. Yu, L.C. Wang, Q.J. Xing, et al., Chin. Chem. Lett. 31 (2020) 1648-1653. DOI:10.1016/j.cclet.2019.08.020 |
| [12] |
K. Qi, N. Cui, M. Zhang, et al., Chemosphere 272 (2021) 129953. DOI:10.1016/j.chemosphere.2021.129953 |
| [13] |
M.Q. Yang, M.M. Gao, M.H. Hong, G.W. Ho, Adv. Mater. 30 (2018) 1802894. DOI:10.1002/adma.201802894 |
| [14] |
Y. Wang, F. Silveri, M.K. Bayazit, et al., Adv. Energy Mater. 8 (2018) 1801084. DOI:10.1002/aenm.201801084 |
| [15] |
J. Li, X.Y. Wu, W.F. Pan, et al., Angew. Chem. Int. Ed. 57 (2018) 491-495. DOI:10.1002/anie.201708709 |
| [16] |
B. Sun, P. Qiu, Z. Liang, et al., Chem. Eng. J. 406 (2021) 127177. DOI:10.1016/j.cej.2020.127177 |
| [17] |
Q.Y. Tian, W.J. Yao, W. Wu, C.Z. Jiang, Nanoscale Horiz. 4 (2019) 10-25. DOI:10.1039/c8nh00154e |
| [18] |
X. Xie, Q. Hassan, H. Lu, et al., Chin. Chem. Lett. 32 (2021) 2038-2042. DOI:10.1016/j.cclet.2020.10.002 |
| [19] |
Y. Qin, Y. Guo, Z. Liang, et al., Chin. Chem. Lett. 32 (2021) 1523-1526. DOI:10.1016/j.cclet.2020.10.020 |
| [20] |
W.C. Huang, Y. Gao, J.X. Wang, et al., Small (2020) 2004557. DOI:10.1002/smll.202004557 |
| [21] |
O. Guselnikova, A. Trelin, E. Miliutina, et al., Acs Appl. Mater. Interfaces 12 (2020) 28110-28119. DOI:10.1021/acsami.0c04029 |
| [22] |
L.W. Wang, X. Zhang, X. Yu, et al., Adv. Mater. 31 (2019) 1901965. DOI:10.1002/adma.201901965 |
| [23] |
Y. Xia, B. Cheng, J. Fan, et al., Sci. China Mater. 63 (2020) 552-565. DOI:10.1007/s40843-019-1234-x |
| [24] |
S. Bellamkonda, R. Shanmugam, R.R. Gangavarapu, J. Mater. Chem. A 7 (2019) 3757-3771. DOI:10.1039/c8ta10580d |
| [25] |
P.J. Yang, R.R. Wang, M. Zhou, X.C. Wang, Angew. Chem. Int. Ed. 57 (2018) 8674-8677. DOI:10.1002/anie.201804996 |
| [26] |
Y.L. Yang, S.C. Wang, Y.L. Jiao, et al., Adv. Funct. Mater. 28 (2018) 1805698. DOI:10.1002/adfm.201805698 |
| [27] |
Y. Huang, D. Li, Z. Fang, et al., Appl. Catal. B: Environ. 254 (2019) 128-134. DOI:10.1016/j.apcatb.2019.04.082 |
| [28] |
Y.Y. Kang, Y.Q. Yang, L.C. Yin, et al., Adv. Mater. 27 (2015) 4572-4577. DOI:10.1002/adma.201501939 |
| [29] |
D.M. Zhao, C.L. Dong, B. Wang, et al., Adv. Mater. 31 (2019) 1903545. DOI:10.1002/adma.201903545 |
| [30] |
J. Tian, D. Wang, S.T. Li, et al., Acs Sustain. Chem. Eng. 8 (2020) 594. DOI:10.1021/acssuschemeng.9b06134 |
| [31] |
Y.X. Li, S.X. Ouyang, H. Xu, et al., J. Am. Chem. Soc. 138 (2016) 13289-13297. DOI:10.1021/jacs.6b07272 |
| [32] |
J.J. Tian, L.X. Zhang, M. Wang, et al., Appl. Catal. B: Environ. 232 (2018) 322-329. DOI:10.1016/j.apcatb.2018.03.076 |
| [33] |
H.G. Yu, H.Q. Ma, X.H. Wu, et al., Solar RRL (2020) 2000372. |
| [34] |
Y.X. Li, H. Xu, S.X. Ouyang, et al., J. Mater. Chem. A 4 (2016) 2943. DOI:10.1039/C5TA05128B |
| [35] |
Y.S. Xu, X. He, H. Zhong, et al., Appl. Catal. B: Environ. 246 (2019) 349-355. DOI:10.1016/j.apcatb.2019.01.069 |
| [36] |
Y.Y. Zhang, Z.X. Zhou, Y.F. Shen, et al., ACS Nano 10 (2016) 9036-9043. DOI:10.1021/acsnano.6b05488 |
| [37] |
J. Oh, R.J. Yoo, S.Y. Kim, et al., Chem. Eur. J. 21 (2015) 6241-6246. DOI:10.1002/chem.201406151 |
| [38] |
S. Wu, H. Yu, S. Chen, X. Quan, ACS Catal 10 (2020) 14380-14389. DOI:10.1021/acscatal.0c03359 |
| [39] |
H. Wang, X.S. Sun, D.D. Li, et al., J. Am. Chem. Soc. 139 (2017) 2468-2473. DOI:10.1021/jacs.6b12878 |
| [40] |
Y.J. Zhang, T. Mori, J.H. Ye, M. Antonietti, J. Am. Chem. Soc. 132 (2010) 6294-6295. DOI:10.1021/ja101749y |
| [41] |
M. Sachs, R.S. Sprick, D. Pearce, et al., Nat. Commun. 9 (2018) 4968. DOI:10.1038/s41467-018-07420-6 |