 2021, Vol. 32
2021, Vol. 32 


b School of Environmental Science and Engineering, Tianjin University, Tianjin 300350, China;
c School of Science, Tibet University, Lhasa 850000, China
Fluoride pollution in groundwater is regarded as a global environmental problem threatening human health. World Health Organization (WHO) standardized the fluoride concentration in drinking water should not exceed 1.5 mg/L. To guarantee this standard, various technologies, included co-precipitation [1], membrane separation [2], electrodialysis [3] and adsorption [4, 5], are developed to purify fluoride-containing water bodies. Adsorption was considered as the most promising method due to its low cost, easy operation, and high efficiency, especially at a low fluoride concentrations [6].
Various adsorbents had been applied to remove fluoride in aqueous solutions, such as zirconium oxide [7], aluminum oxide [8], iron oxide [9], layered double hydroxides (LDHs) [10], and metal-organic frameworks (MOFs) [11] etc. It should be noted that Zr(IV)−based adsorbents, including zirconium oxide [12], hydrous zirconium oxide [7], and Zr−MOFs [13] etc., were a kind of widely popular remediation materials for fluoride. Zr(IV) featured a 4d° configuration acted as Lewis acid, endowing its strong coordination ability to fluoride by Lewis acids−bases interactions. However, most of zirconium-based adsorbents applied for fluoride removal were in powder form (nanoscale or a few to dozens of microns), which were detrimental to solid-liquid separation, regeneration, and fixed-bed adsorption process [14-16]. Thus, millimeter-scale supported adsorbents had been developed to solve the problems. Representative supports include activated carbon [17], ion exchange resin [10], natural polymer [18] and so on. Whereas, internal channel of the supports were easily blocked after loading the zirconium-based adsorbents, which impeded the liquid-solid mass transfer process between fluoride ion and the interface of adsorbent. In addition, due to the poor interaction between these supports and the active components of adsorbent, there was a possibility that the inside adsorbent may fall off from the supports.
When sodium alginate sol was dropped into Zr4+ ion solution, millimeter-sized hydrogel beads with 3D network structure inside were fabricated via ion-crosslinking between Zr4+ ion and monomer structure of β-D mannuronic acid (G) in alginate [19, 20]. The adsorbent prepared by this method was completely different from the supported zirconium-based adsorbents mentioned above, by which a single phase adsorbent of Zr4+ ion−crosslinked alginate hydrogel beads (AHB@Zr) was produced. The removal of fluoride was realized via ion exchange of fluoride ion with hydroxyl group on the zirconium site of AHB@Zr. AHB@Zr was expected to show excellent fluoride removal performance, however, it was difficult to be applied in actual water due to its low adsorption capacity and narrow pH applicability. In order to enhance the ability of fluoride removal, recent studies have shown that sulfate anion could serve as active group on adsorbents for the removal of fluoride ion. For instance, Chai et al. reported that sulfate-doped Fe3O4/Al2O3 nanoparticles was a novel adsorbent to remove fluoride ion from drinking water [21]. Jia et al. prepared sulfate-treated porous 2-line ferrihydrite/bayerite composites (LFBC) [22] and Chen et al. synthesised sulfate-doped hydroxyapatite [23], all of them improved fluoride removal performance. Therefore, introducing sulfate group into the AHB@Zr would be an effective method to improve the adsorption performance of fluoride, unfortunately, this research has not yet been reported.
In this work, sulfate group was anchored on the millimeter-scale Zr4+ ion-crosslinked alginate hydrogel beads (denoted as AHB@Zr-SO42−) via mild sol-gel process (Scheme 1). The effects of pH, coexisting anions, adsorption kinetics, and initial concentrations on fluoride ion removal performance were studied. And, the column adsorption and cycle performance experiments were carried out to evaluate the potential for practical application. The adsorption mechanism was discussed by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR) spectrometer analysis combined with ion chromatography measurement. In order to understand the mechanism of adsorption at the molecular level, adsorption energies were analyzed based on density functional theory (DFT) calculations.

|
Download:
|
| Scheme 1. The synthesis route of AHB@Zr−SO42− composites. | |
{kind=link}
In order to determine the morphology and structure of the synthesized sample, scanning electron microscope (SEM), FTIR Spectrometer and X-ray diffraction (XRD) were employed here. As could be seen from Figs. S1a and b (Supporting information), their surface possessed highly developed macropores that were conducive to expose more active sites and mass transfer. In addition, the results of element mapping analysis indicated that the sulfur element was successfully riveted on the AHB@Zr. The specifics areas and pore size of AHB@Zr and AHB@Zr-SO42− were tested by N2 adsorption-desorption isotherms. The results (Fig. S2 in Supporting information) showed that both of them were type IV with H3 type of hysteresis loop, indicating that the material possessed mesoporous structure. The pore sizes of AHB@Zr and AHB@Zr-SO42− were 3.76 nm and 2.86 nm, and the specific surface area were 7.28 m2/g and 2.52 m2/g, respectively. The decrease of specific surface area of AHB@Zr-SO42− may be due to the effect of the ion-crosslinking between Zr4+ ion and monomer structure of β-D mannuronic acid (G) in alginate when introduction of sulfuric acid. At the same time, we also compared the BET specific surface area of the adsorbent synthesized at different zirconium ion concentrations (Table S1 in Supporting information). We found that the specific surface area decreased when the zirconium ion concentration exceeded 0.2 mol/L. This is because excessive hydrogen ions destroyed the gel of zirconium alginate. This result is also consistent with that the specific surface area of AHB@Zr-SO42− is lower than that of AHB@Zr.
The information of the functional groups on the surface of AHB@Zr and AHB@Zr−SO42− were represented by FTIR spectroscopy. For two materials, absorption peaks at 3416 cm−1 and 1077 cm−1 were ascribed to the stretching vibration of hydroxyl group (Fig. S1c in Supporting information), indicating the presence of adsorption water or crystallization water [24]. Strong sharp peaks appeared at 1665 cm−1 and 1412 cm−1 were assigned to asymmetric stretching vibration and symmetric stretching vibration of carboxyl group, respectively. For AHB@Zr and AHB@Zr−SO42−, the movement from 1665 cm−1 to 1654 cm−1 indicated that Zr4+ was cross-linked with carboxyl group of guluronic acid in alginate sodium. Meanwhile, strong peaks at 1125 cm−1 and 1027 cm−1 were assigned to the sulfate ion of AHB@Zr-SO42− [25], indicating that a coordination bond was formed between sulfate and zirconium sites in AHB@Zr-SO42−. Interestingly, the peak intensity increased with the increasing of the molar ration of SO42− to Zr4+ from 0:1 to 2:1, while the peak intensity decreased in the range of 2:1–4:1, which was probably because that the excessive hydrogen ion destroyed the binding ability between sulfate and zirconium sites. It could be found that the peaks of carboxyl group returned to the previous position. With the increase of sulfate ion concentration, the peak intensity of carboxyl functional group becomes weaker, because the excessive acid destroys the gel structure of zirconium alginate. Besides, we could see from the results of XRD in Fig. S1d (Supporting information) that the material was amorphous [26]. This was related to the aperiodic structure of sodium alginate.
The mechanism of adsorbent synthesis was confirmed by XPS analysis. Survey and high resolution XPS spectra of C 1s, Zr 3d and O 1s were depicted in Fig. 1. The sodium ion on sodium alginate (SA) disappeared because the zirconium ion replaced the sodium ion of SA to achieve the crosslinking of zirconium ion with carboxyl mentioned above. For AHB@Zr-SO42−, the appearance of S 2p characteristic peak further confirmed the formation of coordination bond of SO42− with Zr4+ on the surface of AHB@Zr. C 1s spectra of SA was convoluted into three peaks at 284.6 eV, 286.2 eV, and 288.1 eV (Fig. 1b), which were assigned to C−C, C−O−C and O−C=O, respectively [27]. Binding energy of O−C=O in SA increased from 288.1 eV to 288.5 eV than that in AHB@Zr [28], which was due to the shift of the electron cloud density after zirconium ion cross-linked with carboxyl group (Fig. 1c). After being modified by SO42−, the binding energy of carboxyl group was increased by 0.1 eV, suggesting that a coordination bond (Zr-SO42−) was formed (Fig. 1d). And O 1s spectra was convoluted into three peaks at 530.5 eV, 531.9 eV and 533.0 eV, which assigned to Zr−O, C=O, and C−O, respectively. Moreover, a new peak appeared at 532.4 eV after modified by SO42−, which corresponded to S−O. This could further confirm that sulfate was introduced into the surface of AHB@Zr [16]. Fig. 1f shows that the binding energies of Zr 3d5/2 and Zr 3d3/2 increased from 182.6 eV and 184.9 eV to 183.0 eV and 185.5 eV after being modified by SO42− [29], which indicated that the electron cloud density on the Zr decreased after the Zr4+ connected to SO42−.

|
Download:
|
| Fig. 1. XPS survey spectra of sodium alginate, AHB@Zr and AHB@Zr-SO42− (a), high resolution XPS spectra C 1s spectra of sodium alginate (b), AHB@Zr (c), AHB@Zr−SO42− (d), O 1s (e), Zr 3d (f) spectra of AHB@Zr and AHB@Zr-SO42−. | |
{kind=link}
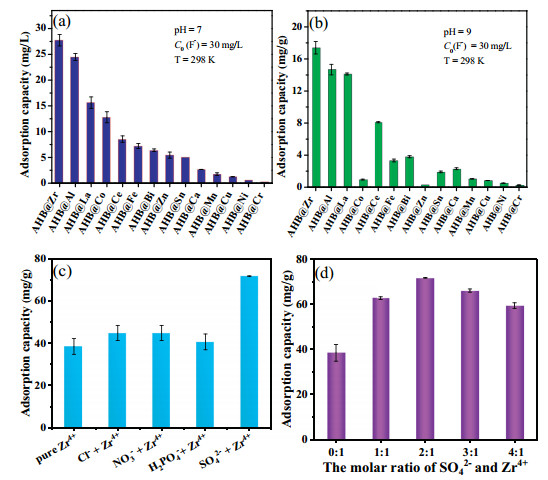
In order to determine the best adsorbent, we optimized the adsorbent gradually. Fourteen kinds of alginate hydrogel beads (AHB@M) (M = Zr4+, Al3+, Fe3+, La3+, Ce3+, Cr3+, Mn2+, Sn4+, Cu2+, Co2+, Ni2+, Zn2+, Ba2+, or Ca2+ ion) were synthesized to compare their defluorination ability. We could clearly see from Figs. 2a and b that fluoride removal ability of AHB@Zr was much higher than other AHB@Ms at pH 7 and 9. In order to further improve the fluoride removal performance of AHB@Zr and broaden its pH application range, we innovatively introduced sulfate into the surface of zirconium alginate, and formed Zr-SO42− on AHB@Zr-SO42−. At the same time, considering that whether other anions (Cl−, NO3− and H2PO4−) would also improve the fluoride removal performance of AHB@Zr, we replaced sulfuric acid with other acids (HCl, HNO3, and H3PO4) during the preparation process. The results (Fig. 2c) showed that other anionic surface-modified AHB@Zr did not significantly improve the removal of fluoride ion compared to the pure AHB@Zr. This further showed that the sulfate played a unique role of active group in AHB@Zr, which may benefit from the strong binding ability of sulfate and zirconium sites on AHB@Zr-SO42−. But Cl−, NO3− and H2PO4− also increase the fluoride removal performance, because there were still hydrochloric acid, nitric acid and phosphoric acid on the surface of the adsorbent, which are released during the adsorption process to promote the adsorption performance. The adsorption capacity of AHB@Zr-SO42− at different molar ratio of SO42− to Zr4+ were shown in Fig. 2d. The adsorption capacities of AHB@Zr-SO42− increased with the increase of the molar ratios of SO42− to Zr4+ from 0:1 to 2:1. This result showed that sulfate does strengthen the adsorption capacity of AHB@Zr. However, the adsorption capacity declined when the molar ratio of SO42− to Zr4+ exceeded 2:1. The excessive sulfuric acid destroyed the ion-crosslinking process, thus reduced the content of sulfate on AHB@Zr-SO42−, which can be confirmed by the FTIR characterization. Based on this, we determined that the molar ratio of SO42− to Zr4+ in AHB@Zr-SO42− was 2:1.

|
Download:
|
| Fig. 2. Fluoride removal with different metal ions-crosslinking alginate hydrogel beads at pH 7 (a) and pH 9 (b). The adsorption capacity by different anions modified AHB@Zr (c). The effect of molar ratios of SO42− to Zr4+ on fluoride removal (d). | |
{kind=link}
The pH of solution was a very important factor that affecting the species of ions and the surface charge of adsorbent, which determined the adsorption performance. Fig. S3a (Supporting information) shows the adsorption capacities of the two adsorbents at different pH (2.0–11.0). The adsorption capacity of AHB@Zr-SO42− was always higher than that of AHB@Zr. While when pH exceeded 9, the adsorption capacity dropped sharply, this was attributed to the competition of excessive hydroxide ions in the solution with fluoride ions. In order to verify the effect of surface charge on fluoride removal behavior of AHB@Zr and AHB@Zr-SO42−, we tested the point of zero charge (pHpzc) of the two adsorbents. It was observed that the pHpzc of AHB@Zr-SO42− was 6.71, while the pHpzc of AHB@Zr was 4.78 (Fig. S3b in Supporting information). For AHB@Zr, the adsorption process was favorable when pH < pHPZC [30], hydroxyl group on its surface was protonated with positive charge, thus the fluorine ion in solution could be adsorbed by electrostatic attraction (Eqs. 1 and 2). In addition, hydroxyl group on the surface of AHB@Zr would undergo an ion exchange process with F− (Eq. 3). For AHB@Zr-SO42−, the SO42− on the its surface would also undergo an ion-exchange process with F− (Eq. 4). However, when pH < 2, F− existed in the form of HF which reduced the adsorption efficiency to some extent. In particular, the Eqs. 1–3 were susceptible to pH variation, while the Eq. 4 was unaffected. This was why the pH applicability of AHB@Zr-SO42- was broader than AHB@Zr. In actual fluoride-containing groundwater, there were some co-existing anions (Cl−, SO42−, NO3−, HCO3− and PO43−) that would compete with fluoride for adsorption sites. So we investigated the effect of co-existing anions on fluoride adsorption performance and the results were shown in Fig. S4 (Supporting information).
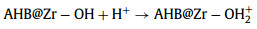
|
(1) |
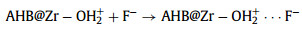
|
(2) |

|
(3) |

|
(4) |
In order to explore the saturated adsorption amount and adsorption characteristics, the adsorption isotherms of AHB@Zr and AHB@Zr-SO42− were conducted. The parameters fitted by Langmuir [31] and Freundlich models were listed in Table S2 (Supporting information). We could clearly see that the maximum adsorption capacity of AHB@Zr was ~40 mg/g, which was much higher than previously reported porous zirconium alginate adsorbents [32]. The improvement of adsorption performance might be attributed to the enhanced interfacial mass transfer by confined water in the pore channels of AHB@Zr. We further introduced sulfate on AHB@Zr, and as expected, the adsorption capacity of AHB@Zr improved greatly (Fig. S5 in Supporting information). The adsorption capacity of AHB@Zr-SO42− (101.3 mg/g) was ~2.5 times that of AHB@Zr, which were better than most previously reported adsorbents (Table S3 in Supporting information). More importantly, unlike frequently reported powder adsorbents, AHB@Zr-SO42− was a millimeter level in appearance, which not only facilitated solid-liquid separation but also could be used in fixed bed adsorption. Temperature could affect the interface adsorption-desorption process and promote or inhibit the removal performance of F−. While we found that when the reaction temperatures were (25 ℃, 35 ℃ and 45 ℃), it had no effect on the adsorption of F−. In actual application process, the kinetics of F− removal were important for selecting the optimum operating conditions. As shown in Fig. S6 (Supporting information), only after 4 h, both adsorbents reached equilibrium. At the same time, we performed a fitting analysis on the adsorption kinetics. It could see that the R2 value of the pseudo-second-order model was higher than the pseudo-first-order kinetic model, indicating a chemical adsorption process, and the fitting parameters were showed in Table S4 (Supporting information).
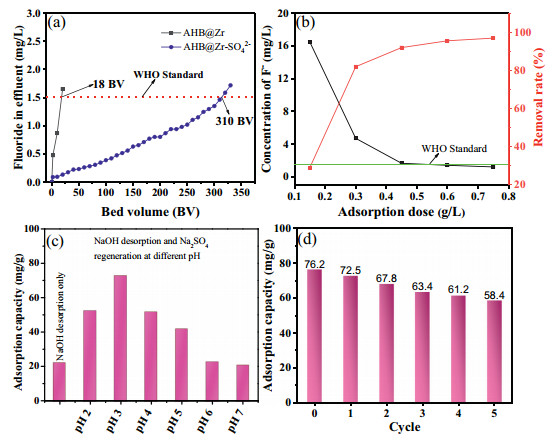
We used AHB@Zr and AHB@Zr-SO42− to conduct fixed bed operation tests respectively in simulated groundwater containing F−. The information of groundwater was as follows: pH 8.0–8.3, F−: 4.1 mg/L, Cl−: 74.6 mg/L, SO42−: 67.2 mg/L, HCO3−: 20.1 mg/L, NO3−: 16.7 mg/L, total P: 0.11 mg/L. As shown in Fig. 3a, the processing capacity of AHB@Zr-SO42− was 310 bed volumes (BV), which was far more than AHB@Zr (~15 BV). In addition, we tested the Cl−, SO42−, HCO3−, NO3−, total P concentrations in the solution when the adsorbent reached the adsorption saturation state, and the results are listed in Table S5 (Supporting information). We can find that the concentration of Cl− and NO3− basically did not change, but the concentrations of total P and HCO3− reduced significantly. This result is also consistent with the results of the competitive ion experiment in the static adsorption experiment. The increase in SO42− concentration is due to ion exchange between the adsorbent and fluoride ion. For a batch adsorption process, the optimal dosage could not only ensure the effective removal of fluoride, but also reduced the cost of adsorbent. So we investigated the effect of AHB@Zr-SO42− (0.1–0.8 g/L) dosages on fluoride removal performance and the results were showed in Fig. 3b. It could see that an increase of AHB@Zr-SO42− dosages from 0.1 g/L to 0.8 g/L had positive effect on fluoride removal and the removal rate increased from 45.29% to 97.21%, this could be attributed to the increase of adsorption sites. And it could meet WHO drinking water standard (below 1.5 mg/L) when AHB@Zr-SO42− dosage reached above 0.53 g/L (C0 = 30 mg/L). Moreover we used AHB@Zr-SO42− to treat the actual fluorine-containing surface water in western China (pH 9, Ci = 15.83 mg/L), the results were shown in Fig. S7 (Supporting information). When the dose of the adsorbent was greater than 0.65 g/L, the fluoride ion concentration in the actual fluoride-containing surface water could be reduced to below 1.0 mg/L. Furthermore, column adsorption experiment was conducted on this actual fluoride-containing surface water. As shown in Fig. S7, AHB@Zr-SO42− column was able to generate less than ~26 BV of purified water ([F] < 1.0 mg/L) from F− contaminated water successively.

|
Download:
|
| Fig. 3. Column adsorption study of fluoride by using AHB@Zr-SO42− (a), effects of adsorbent dose for removal rate (b), optimization of desorption process (c), reusability of AHB@Zr-SO42− (d). | |
{kind=link}
The reusability of the adsorbent was an important aspect to evaluate its potential in practical application. As shown in Fig. 3c, only NaOH was used for desorption, the adsorption performance was only 20% of the fresh adsorbent. This was due to the consumption of sulfate ion during adsorption process but no supplementation. So we used sodium sulfate solution to regenerate the adsorbent after being desorbed with sodium hydroxide. Furthermore, the pH of sodium sulfate solution had significant effect on the defluoridation performance of the regenerated adsorbent. When sodium sulfate was in acidic environment, it was beneficial to the regeneration of adsorbent, this might be attributed to the coordination of SO42− with Zr4+ under acidic conditions, and the results were consistent with the preparation process of SO42− modified AHB@Zr. The adsorption performance of the regenerated adsorbent by sodium sulfate solution (pH 3) could reach 95% of the fresh adsorbent, and after five cycles of desorption-regeneration, the defluorination performance of adsorbent still maintained above 80%, this meant that the adsorbent had good reproducibility (Fig. 3d).
The adsorption mechanism of AHB@Zr-SO42− was studied by using FTIR analysis. The FTIR spectrum of AHB@Zr-SO42− adsorbed by different concentrations of fluoride (25 mg/L, 50 mg/L, 75 mg/L, 100 mg/L) and the results were showed in Fig. 4a. As the concentration of F− increased, the peak intensity gradually disappeared, indicating that the adsorption mechanism was ion exchange between SO42− and F−. At the same time, we also monitored the concentration of F− and SO42− in the solution during adsorption process, and the results were showed in Fig. 4b. We found that the concentration of F− decreased with increasing the concentration of SO42−. After reaching adsorption equilibrium, the molar concentration ratio of adsorbed F− and released SO42− was about 2:1, which could effectively prove that the removal mechanism was ion exchange between SO42− and F−.

|
Download:
|
| Fig. 4. FTIR spectra of AHB@Zr-SO42− after adsorption (a), changes in the concentration of SO42− and F− with time (b), XPS F 1s spectrum of the AHB@Zr-SO42− after adsorption (c), S 2p spectrum of the AHB@Zr-SO42− before and after fluoride adsorption (d), XPS Zr 3d spectra of the AHB@Zr-SO42− before and after fluoride adsorption (e). | |
{kind=link}
XPS analysis was used to study the defluorination mechanism of AHB@Zr-SO42−. As shown in Figs. 4c–e. After AHB@Zr-SO42− adsorbed F−, F 1s spectrum appeared and S 2p spectrum disappeared, indicating the adsorption mechanism was ion exchange between SO42− and F−. After fluoride adsorption, the binding energy of Zr 3d did not change significantly, which meant that the electron cloud density around Zr had no significantly change when F− replaced SO42−. The defluoridation mechanism of AHB@Zr was further confirmed by XPS analysis. Fig. S8 (Supporting information) showed O 1s spectra was convoluted into three peaks at 530.7 eV, 532.2 eV, and 533.3 eV, which assigned to Zr−O, Zr−OH and C−O, respectively. After AHB@Zr adsorbed F−, the peak area of Zr−OH decreased by 11.9% (Fig. S8), which proved that the adsorption mechanism is ion exchange between the hydroxyl on the zirconium site and fluoride ions. As shown in Fig. S9 (Supporting information) there was no obvious change in the structure of AHB@Zr-SO42− before and after adsorption, indicating that AHB@Zr-SO42− had good stability. Moreover, it could see from the energy dispersive X-Ray spectroscopy (EDS) spectrum that sulfur element disappeared basically after the adsorption of F− by AHB@Zr-SO42−, which again proved that the defluorination mechanism was ion exchange.
Density functional theory (DFT) calculations were conducted to study the reason why F− removal performance of AHB@Zr-SO42− was better than AHB@Zr and to study the energy changes before and after adsorption at molecular level. The results of DFT calculations displayed that the binding energy of AHB@Zr during F− removal process was −1.09 eV, the negative value meant that the process of F− removal by AHB@Zr was thermodynamically feasible. F− attacked the chemical bond between zirconium and hydroxyl group (i.e., Zr−OH) and further to form a stronger zirconium fluoride bond (Zr−F). The binding energy of AHB@Zr-SO42− during F− removal process was −3.80 eV, it was more negative than the binding energy value of AHB@Zr, which meant a more thermodynamically feasible process (Fig. S10 in Supporting information). Therefore, F− was easier to exchange with SO42− on AHB@Zr-SO42−than AHB@Zr.
In this work, AHB@Zr−SO42− was fabricated via introducing sulfate groups to zirconium sites on the surface of AHB@Zr to realize 3D porous structure with abundant macropores and mesopores. Thus, its stable defluorination ability in the wide pH range of 3–9 was better than that of AHB@Zr, which was due to the difference of adsorption energy between AHB@Zr-SO42− and AHB@Zr (−3.18 eV vs. −1.09 eV). The maximum adsorption capacity of AHB@Zr-SO42− was up to 101.3 mg/g, this was significantly better than other millimeter-scale and some nano-scale adsorbents. More importantly, the exhausted adsorbent could be used via desorbtion−regeneration to realize reuse. After 5 cycles, the adsorption capacity could be maintained above 80%. By means of characterization analysis and DFT calculation, we proved the mechanism of fluoride removal was ion exchange between F− and SO42−. This provided a new strategy for the preparation of low-cost and high-capacity bio-based adsorbents.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the Natural Science Foundation of Tianjin (No. 18JCYBJC17700), the National Natural Science Foundation of China (Nos. 21406164, 21466035 and 22066022).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.04.057.
| [1] |
C.J. Huang, J.C. Liu, Water Res. 33 (1999) 3403-3412. DOI:10.1016/S0043-1354(99)00065-2 |
| [2] |
M. Tahaikt, R. El Habbani, A.A. Haddou, et al., Desalination 212 (2007) 46-53. DOI:10.1016/j.desal.2006.10.003 |
| [3] |
Z. Amor, B. Bariou, N. Mameri, et al., Desalination 133 (2001) 215-223. DOI:10.1016/S0011-9164(01)00102-3 |
| [4] |
X. Wu, X. Tan, S. Yang, et al., Water Res. 47 (2013) 4159-4168. DOI:10.1016/j.watres.2012.11.056 |
| [5] |
W. Qiao, P. Zhang, L. Sun, et al., Chem. Lett. 31 (2020) 2742-2746. |
| [6] |
S. Jagtap, M.K. Yenkie, N. Labhsetwar, et al., Chem. Rev. 112 (2012) 2454-2466. DOI:10.1021/cr2002855 |
| [7] |
X. Dou, D. Mohan, C.U. Pittman, S. Yang, et al., Chem. Eng. J. 198-199 (2012) 236-245. DOI:10.1016/j.cej.2012.05.084 |
| [8] |
S. Ayoob, A.K. Gupta, P.B. Bhakat, et al., Chem. Eng. J. 140 (2008) 6-14. DOI:10.1016/j.cej.2007.08.029 |
| [9] |
A.A. Markeb, A. Alonso, A. Sánchez, et al., Sci. Total Environ. 598 (2017) 949-958. DOI:10.1016/j.scitotenv.2017.04.191 |
| [10] |
J. Cai, Y. Zhang, B. Pan, et al., Water Res. 102 (2016) 109-116. DOI:10.1016/j.watres.2016.06.030 |
| [11] |
Q. Gao, J. Xu, H. X., et al., Coord. Chem. Rev. 378 (2019) 17-31. DOI:10.1016/j.ccr.2018.03.015 |
| [12] |
Z. Ren, G. Zhang, J.P. Chen, et al., J. Colloid Interface Sci. 358 (2011) 230-237. DOI:10.1016/j.jcis.2011.01.013 |
| [13] |
J. He, X. Cai, K. Chen, Y. Li, et al., J. Colloid Interface Sci. 484 (2016) 162-172. DOI:10.1016/j.jcis.2016.08.074 |
| [14] |
T.L. Tan, P.A.P. Krusnamurthy, H. Nakajima, et al., RSC Adv. 10 (2020) 18740-18752. DOI:10.1039/D0RA01268H |
| [15] |
L. Mei, C. Peng, H. Qiao, et al., RSC Adv. 9 (2019) 33345-33353. DOI:10.1039/C9RA07155E |
| [16] |
J. He, T.S. Siah, J.P. Chen, et al., Water Res. 56 (2014) 88-97. DOI:10.1016/j.watres.2014.02.030 |
| [17] |
S. Bakhta, Z. Sadaoui, U. Lassi, et al., Chem. Phys. Lett. 754 (2020) 137705. DOI:10.1016/j.cplett.2020.137705 |
| [18] |
Y. Zhang, S. Lin, J. Qiao, et al., Chem. Eng. J. 353 (2018) 225-236. DOI:10.1016/j.cej.2018.06.143 |
| [19] |
Y. Cheng, L. Lu, W. Zhang, et al., Carbohydr. Polym. 88 (2012) 1093-1099. DOI:10.1016/j.carbpol.2012.01.075 |
| [20] |
H. Luo, X. Zeng, P. Liao, et al., Int. J. Biol. Macromol. 126 (2019) 1133-1144. DOI:10.1016/j.ijbiomac.2018.12.269 |
| [21] |
L. Chai, Y. Wang, N. Zhao, et al., Water Res. 47 (2013) 4040-4049. DOI:10.1016/j.watres.2013.02.057 |
| [22] |
Y. Jia, B.S. Zhu, K.S. Zhang, et al., Chem. Eng. J. 268 (2015) 325-336. DOI:10.1016/j.cej.2015.01.080 |
| [23] |
L. Chen, K.S. Zhang, J.Y. He, et al., Chem. Eng. J. 285 (2016) 616-624. DOI:10.1016/j.cej.2015.10.036 |
| [24] |
H. Niu, Q. Yang, K. Tang, et al., J. Nanopart. Res. 8 (2006) 881-888. DOI:10.1007/s11051-006-9138-x |
| [25] |
J. He, Y. Li, X. Xue, et al., RSC Adv. 6 (2016) 43814-43822. DOI:10.1039/C6RA03590F |
| [26] |
B. Pan, J. Xu, B. Wu, et al., Environ. Sci. Technol. 47 (2013) 9347-9354. DOI:10.1021/es401710q |
| [27] |
H. Viltres, O.F. Odio, L. Lartundo-Rojas, et al., Appl. Surf. Sci. 511 (2020) 145606. DOI:10.1016/j.apsusc.2020.145606 |
| [28] |
H. Ao, W. Cao, Y. Hong, et al., Total Environ. 708 (2020) 135092. DOI:10.1016/j.scitotenv.2019.135092 |
| [29] |
Z. Yu, C. Xu, K. Yuan, et al., J. Hazard. Mater. 346 (2018) 82-92. DOI:10.1016/j.jhazmat.2017.12.024 |
| [30] |
H. Zhang, Y. Ruan, Y. Feng, et al., J. Colloid Interface Sci. 536 (2019) 180-188. DOI:10.1016/j.jcis.2018.10.054 |
| [31] |
S.P. Wu, X.Z. Dai, et al., Chin. Chem. Lett. 28 (2017) 625-632. DOI:10.1016/j.cclet.2016.11.015 |
| [32] |
Q. Zhou, X. Lin, J. Qian, et al., RSC Adv. 5 (2015) 2100-2112. DOI:10.1039/C4RA12036A |