 2021, Vol. 32
2021, Vol. 32 


b Department of Biomedical Science, Acharya Narendra Dev College, University of Delhi, India;
c Department of Chemistry, University of Massachusetts Boston, 100 Morrissey Boulevard, Boston, MA 02125, United States;
d Hamari Chemical Ltd., 1-4-29 Kunijima, Higashi-Yodogawa-ku, Osaka 533-0024, Japan;
e Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, 20018 San Sebastián, Spain;
f IKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, Plaza Bizkaia, 48013 Bilbao, Spain
Due to the growing impact of fluorinated compounds on the progress of energy [1], food [2], and health [3] related industries, organofluorine chemistry has become a multidisciplinary research area of general scientific interest. Indeed, virtually every organic chemistry research group has a certain interest in either synthesis or application of fluorinated molecules. Consequently, there has been an upsurge in covering the wealth of fluorine chemistry by critical thematic reviews focusing on various facets of fluorine, including new reagents [4], synthesis [5], biological properties and applications [6]. Of particular interest to the practitioners is the application of fluorinated compounds in the design of modern pharmaceuticals [3, 7]. This multidisciplinary area of fluorine chemistry is constantly undergoing rapid development requiring timely updates to properly disseminate the new results, ideas, and methodological breakthroughs. To address this critical information need, we have initiated a series of mini-review focusing on the very recent entries of fluorine-containing drugs to the pharmaceutical market. The reviews covering fluorine-containing drugs approved by the US FDA in 2018[8] and 2019 [9] were highly appreciated by the readers, as it follows from the number of downloads and citations. In the present review, we profile 13 new fluorinated drugs approved by the FDA last year (2020) for commercial use. The compounds discussed in the present work include (Fig. 1) AyvakitTM (1), IsturisaTM (2), KoselugoTM (3), PemazyreTM (4), TabrectaTM (5), QinlockTM (6), Nurtec ODTTM (7), CeriannaTM (8), TauvidTM (9), InqoviTM (10), PralsebinibTM (11), OrladeyoTM (12), OrgovyxTM (13). Most of the compounds were developed to treat various cancers (1, 4-6, 8, 11, 13). Other therapeutic areas include Cushing's disease (2), neurofibromatosis (3), migraine (7), Alzheimer's disease (9), myelodysplastic syndromes (10), and hereditary angioedema (11). For each compound, we discuss the medicinal chemistry discovery, and biological activity, emphasizing synthesis and introduction of fluorine.

|
Download:
|
| Fig. 1. Fluorine-containing drugs approved by the US FDA in 2020. | |
{kind=link}
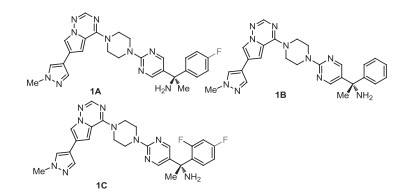
Avapritinib (1), also named BLU-285, was developed by Blueprint Medicines as a potent type Ⅰ inhibitor of KIT and platelet-derived growth factor receptor alpha (PDGFRA) activation loop mutants. It is a chiral compound containing one (S)-configuration quaternary carbon center and a mono-fluorinated phenyl moiety (Fig. 1). The structure-activity relationship (SAR) study by Blueprint Medicines disclosed that avapritinib (1) showed the best activity [10] with PDGFRA D842V (half maximal inhibitory concentration, IC50 = 0.24 nmol/L), and KIT exon 11 (IC50 0.6 nmol/L). Structural variations, such as opposite configuration (1A), non-fluoro substituent (1B) and introduction of two fluoro atom (1C), led to the decreased activities (Fig. 2). Furthermore, avapritinib (1) was a highly selective inhibitor of KIT and PDGFRA activation loop mutant comparing with other compounds, such as imatinib, sunitinib and regorafenib with IC50 value (PDGFRA exon 18) of 759 nmol/L, 120 nmol/L, and 810 nmol/L, respectively [11]. Avapritinib was approved in the USA in January 2020, with the trade name AyvakitTM, which was used to treat gastrointestinal stromal tumor (GIST) harboring a PDGFRA exon 18 mutation, including PDGFRA D842V mutations [12].

|
Download:
|
| Fig. 2. Structures of 1A, 1B and 1C. | |
{kind=link}
The methods for the synthesis of avapritinib (1) usually rely on the reaction between amine 19 and aryl chloride 20. In 2015, Blueprint Medicines patented a related method for making racemic 1 as the product, which needed separation via chiral supercritical fluid chromatogram (SFC) [10]. In 2020, an asymmetric catalytic method with bis[(2, 3, 5, 6-η)-(1R, 4R)-2, 5-2, 5-bis(4-trifluoromethylphenyl)bicyclo[2.2.2]octane-2-diene]bis-μ-rhodium chloride (16) as catalyst was developed (Scheme 1) [13]. First, condensation reaction between 4-fluoroacetophenone and p-toluenesulfonamide under reflux for 24 h generated imine 14. Imine 14 was subjected to Rh-catalyzed asymmetric addition reaction [14] of potassium 2-(1-piperazinyl)pyrimidine-5-trifluoroborate (15) in the presence of toluene-4-sulfonic acid to give the chiral sulfonamide 17. Deprotection of tosyl group of sulfonamide 17 via treatment with thiophenol and potassium carbonate (K2CO3) afforded the free amine 18, which was protected by Boc-group to produce the compound 19 [15]. Then, substitution reaction between compound 19 and 4-chloro-6-(1-methyl-1H-pyrazol-4-yl)pyrrolo[1, 2-f][1, 2, 4]triazine (20) in the presence of DIPEA at room temperature for 5 h gave the intermediate 21. Finally, intermediate 21 was treated with 4 mol/L HCl in dioxane at room temperature for 1 h to provide the target product avapritinib (1) [13, 15].

|
Download:
|
| Scheme 1. Synthesis of avapritinib (1). | |
{kind=link}
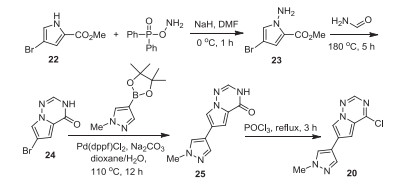
The synthesis of intermediate aryl chloride 20 is shown in Scheme 2 [16]. In the presence of NaH, an amino group was introduced into methyl 4-bromo-1H-pyrrole-2-carboxylate (22) with O-(diphenylphosphoryl)hydroxylamine as the amine source. The resulted intermediate 23 was heated with formamide at 180 ℃ to give cyclic intermediate 24, which underwent the Suzuki coupling reaction with 1-methyl-4-(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl)-1H-pyrazole at 110 ℃ for 12 h. The obtained triazin-4(3H)-one intermediate 25 was chlorinated by treating with POCl3 to afford the key intermediate 20.

|
Download:
|
| Scheme 2. Synthesis of intermediate 20. | |
{kind=link}
3. Osilodrostat (IsturisaTM)
Osilodrostat (2) was developed by Novartis as a potent small-molecule inhibitor of aldosterone synthase (CYP11B2) and 11β-hydroxylase (CYP11B1). Osilodrostat (2) is an asymmetric imidazole-derived compound featuring an essential mono-fluorinated phenyl moiety (Fig. 1) [17]. It was reported that compound FAD286 was a potent CYP11B2 inhibitor [18]. Based on this understanding, Novartis started their SAR investigation and modified the FAD286 (2A) scaffold via variations on the ring size and the substituents on the phenyl group (Fig. 3). Careful SAR study led to the discovery of osilodrostat (2) with an IC50 value of 9 nmol/L against CYP11B2. Fluoro group is also essential for bioactivity. Changing the fluoro group to others, such as chloro (2B), methoxyl, and bromo, decreased the potency against CYP19 [17]. Osilodrostat (2) received its first approval in January 2020 in the European Union (EU) and was approved by US FDA in March with the trade name IsturisaTM for the treatment of Cushing's disease [19].

|
Download:
|
| Fig. 3. Structures of 2A and 2B. | |
{kind=link}
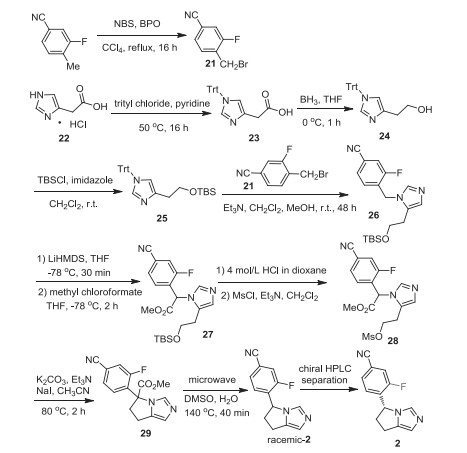
The synthesis of osilodrostat (2) is accessed as showed in Scheme 3 [20]. Treatment of 3-fluoro-4-methylbenzonitrile by N-bromosuccinimide (NBS) in the presence of benzoyl peroxide (BPO) in tetrachloride provided the benzyl bromide intermediate 21. On the other hand, 2-(1H-imidazol-4-yl)acetic acid was protected by trityl group via reaction with trityl chloride in the presence of pyridine, affording the intermediate 23. Then, the carboxylic group of intermediate 23 was reduced by BH3/THF to give alcohol 24 which was protected with butyldimethylsilyl generating intermediate 25. Substitution reaction between benzyl bromide intermediate 21 and intermediate 25 with triethylamine as a base at room temperature for 48 h provided intermediate 26. Deprotonation of intermediate 26 with LiHMDS and followed by reaction with methyl chloroformate in THF at -78 ℃ gave ester 27. Changing the O-protecting group from TBS to methanesulfonyl (Ms) resulted in compound 28, which underwent intramolecular cyclization reaction with the aid of K2CO3 and NaI. Under the microwave heating conditions, decarboxylation of the intermediate 29 at 140 ℃ gave racemic-2, which was separated by chiral HPLC affording the desired osilodrostat (2).

|
Download:
|
| Scheme 3. Synthesis of osilodrostat (2). | |
{kind=link}
Osilodrostat (2) and avapritinib (1) are chiral compounds. Self-disproportionation of enantiomers (SDE) phenomenon may exist during their work-up and isolation procedures in the synthesis, influencing the stereochemical outcome[21]. In this regard, we would like to recommend SDE-tests for accurate determination of stereoselectivity for their asymmetric synthesis by subjecting enantiomerically enriched mixtures into physicochemical processes, such as achiral chromatography [22], evaporation [23], distillation [24] and sublimation [25].
4. Selumetinib (KoselugoTM)Selumetinib (3, AZD6244, ARRY-142886) was developed by AstraZeneca and Merck as an inhibitor of mitogen-activated protein kinase 1 and 2 (MEK1/2) for the treatment of a rare genetic disease called neurofibromatosis, which emerges in early childhood and causes small tumors to grow all over a person's nerves [26-28]. Selumetinib (3) is an N-alkylated benzimidazole derivative (Fig. 1), which was discovered by Array Biopharma in 2003 as a MEK inhibitor. Based on the N-alkylated benzimidazole precursor, Array Biopharma screened many related compounds 30 with modifications on the substituents at different positions (Fig. 4). They chose a dozen of promising compounds exhibiting IC50 (MEK1) value of less than 50 mmol/L [27]. All these selected compounds containing a fluoro substituent at 4-position of benzimidazole clearly indicate the key role of the fluorine atom for the inhibitory activity. Selumetinib (3) was approved by the US FDA in April 2020 under the trade name KoselugoTM for the treatment of tumors associated with neurofibromatosis and various cancers[26].

|
Download:
|
| Fig. 4. Structure of benzimidazole derived MEK inhibitor 30. | |
{kind=link}
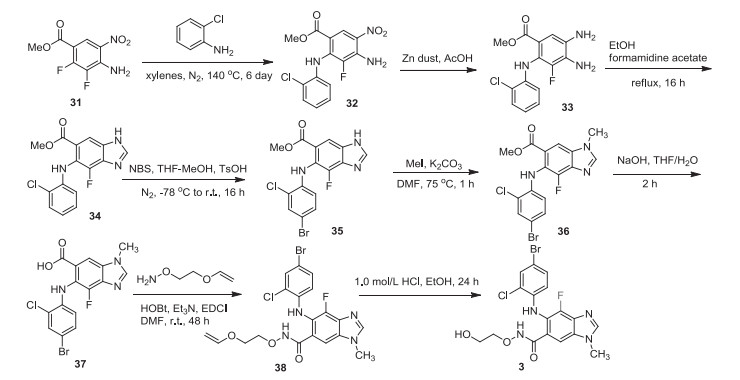
The synthesis of selumetinib (3) is shown in Scheme 4 [27], which was reported by Array Biopharma using methyl 4-amino-2, 3-difluoro-5-nitrobenzoate (31) as the starting material. Intermolecular substitution reaction of fluorinated benzene 31 with 2-chloro aniline in xylenes under the nitrogen atmosphere at 140 ℃ for 6 days generated intermediate 32 which was subjected to reduction reaction with the use of zinc dust in the presence of acetic acid and diamine 33 was obtained. Cyclization reaction of 33 with formamidine acetate under reflux for 16 h gave benzimidazole intermediate 34. The treatment of benzimidazole intermediate 34 with NBS in the presence of TsOH gave brominated intermediate 35. Subsequently, N-methylation of benzimidazole via the reaction with iodomethane in the presence of K2CO3 resulted in intermediate 36, which was subjected to the hydrolysis reaction to give acid 37. The condensation reaction of acid 37 with O-(2-vinyloxy-ethyl)-hydroxylamine was conducted using 1-hydroxybenzotriazole (HOBt) and EDCI in DMF at room temperature for 48 h to give amide 38 in excellent yield (90%). Finally, the desired compound 3 was prepared by deprotection with 1.0 mol/L HCl in ethanol.

|
Download:
|
| Scheme 4. Synthesis of selumetinib (3). | |
{kind=link}
In 2007, Array Biopharma also reported an alternative method for the preparation of intermediate 36, which was obtained by bromination and chlorination reaction of methyl 4-fluoro-5-(phenylamino)-1H-benzo[d]imidazole-6-carboxylate[29].
5. Pemigatinib (PemazyreTM)Pemigatinib (4, INCB054828), developed by Incyte, is a selective fibroblast growth factor receptor (FGFR) inhibitor, which has inhibitory effects on FGFR1 (IC50 0.4 nmol/L), FGFR2 (IC50 0.5 nmol/L), and FGFR3 (IC50 1.0 nmol/L). Pemigatinib (4) shows a weaker inhibitory activity against FGFR4 with an IC50 value of 30 nmol/L. Pemigatinib takes advantage of a hydrophobic selectivity pocket, often using a functionalized dimethoxyphenyl ring to gain FGFR selectivity and enhance the binders' overall potency [30]. Pemigatinib (4) contains tricyclic precursor, 1, 3, 4, 7-tetrahydro-2H-pyrrolo[3′, 2′: 5, 6]pyrido[4, 3-d]pyrimidin-2-one, and a difluoro phenyl moiety (Fig. 1). Incyte also conducted a careful SAR study through the variation of substituents on the phenyl group leading to the discovery of pemigatinib [31]. Pemigatinib was approved by the US FDA in April 2020 under the trade name Pemazyre. It is used for the treatment of locally advanced or metastatic cholangiocarcinoma patients who have received FGFR2 fusion or rearrangement [32].
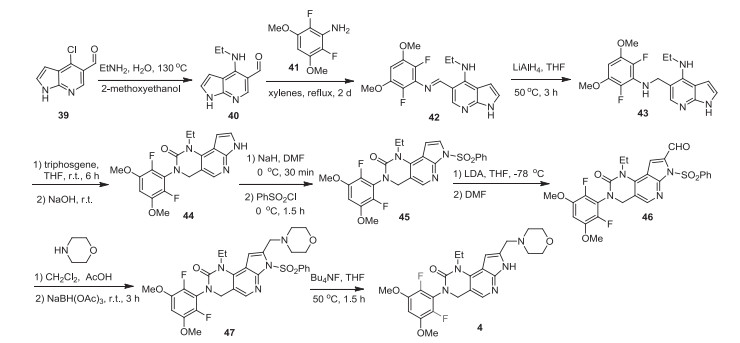
The preparation of pemigatinib (4) developed by Incyte is shown in Scheme 5 [31, 33], which started from a substitution reaction to introduce ethylamino group to 4-chloro-1H-pyrrolo[2, 3-b]pyridine-5-carbaldehyde (39). The generated aldehyde intermediate 40 underwent condensation reaction with 2, 6-difluoro-3, 5-dimethoxyaniline for 2 days to give imine 42, which was subsequently reduced by LiAlH4, affording the amine intermediate 43. Then, the amine intermediate 43 reacted with triphosgene in THF resulting in the cyclization intermediate 44 after 6 h at room temperature. Deprotonation of the intermediate 44 by NaH in DMF followed by reaction with benzenesulfonyl chloride afforded the intermediate 45. Then, formylation of intermediate 45 with the use of LDA and DMF generated aldehyde 46. Reductive amination of aldehyde 46 with morpholine by use sodium triacetoxyborohydride as a reductant, finally, the sulfonyl protecting group was removed via the treatment with tetrabutylammonium fluoride in THF to give pemigatinib (4).

|
Download:
|
| Scheme 5. Synthesis of pemigatinib (4). | |
{kind=link}
6. Capmatinib (TabrectaTM)
Capmatinib (5, INCB 28060) was discovered by Incyte Corporation and developed by Novartis in 2009. Capmatinib is a highly selective and potent inhibitor of the MET receptor tyrosine kinase and has demonstrated clinically meaningful efficacy and a manageable safety profile in patients with advanced non–small-cell lung cancer harboring MET exon 14–skipping mutations [34]. The chemical structure of capmatinib is shown in Fig. 1, which contains an imidazo[1, 2-b][1, 2, 4]triazine key moiety and a mono-fluorinated phenyl group. SAR study by Incyte was mainly based on the two key structural units [35, 36]. Based on results from Phase Ⅱ, capmatinib was approved by the US FDA in May 2020 under the trade name TabrectaTM for the treatment of metastatic non-small cell lung cancer (NSCLC)[37].
The synthesis of capmatinib (5) is illustrated in Scheme 6 [35, 36], which was developed by Incyte. First, a key intermediate, 2-chloro-3-(quinolin-6-yl)propanal (49) was synthesized from the chlorination of 3-(quinolin-6-yl)propanal (48) with NCS. On the other hand, 4-bromo-3-fluoro-N-methoxy-N-methylbenzamide (51) was prepared from 4-bromo-3-fluorobenzoic acid via the formation of acyl chloride and amidation reaction with N, O-dimethylhydroxylamine. Compound 51 was converted into methyl ketone 52 via the reaction with methylmagnesium chloride in THF, which was further treated with HBr and followed by ethyl orthoformate under reflux to afford intermediate 53. Intermediate 53 underwent a cyclization reaction with aminoguanidine bicarbonate in potassium hydroxide (KOH) under reflux resulting in 1, 2, 4-trizazin-3-amine 54. Then, a second cyclization reaction was conducted, and 1, 2, 4-trizazin-3-amine 54 was converted to heterocyclic intermediate 55. Under microwave heating conditions, the Pd-catalyzed coupling reaction between intermediate 55 and zinc cyanide in the presence of N, N, N', N'-tetramethylethylenediamine (TMPDA) gave nitrile intermediate 56, which was transferred into the final capmatinib (5) via hydrolysis under acidic conditions and followed by amidation with methylamine.

|
Download:
|
| Scheme 6. Synthesis of capmatinib (5). | |
{kind=link}
7. Ripretinib (QinlockTM)
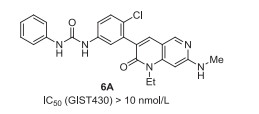
Ripretinib (6, DCC-2618) was developed by Deciphera Pharmaceuticals as a tyrosine kinase inhibitor that inhibits KIT proto-oncogene receptor tyrosine kinase and platelet-derived growth factor receptor A (PDGFRA) kinase. Ripretinib (6) also contains a mono-fluoro substituent on phenyl group, which has been demonstrated to be very important for the inhibitory activity via SAR study (Fig. 1). For example, the IC50 (GIST430) value of ripretinib (6) is less than 10 nmol/L, and ripretinib also shows high activity against GIST48, GIST T1, and GIST882. However, non-fluorinated analog 6A displaces less efficiency against GIST430 with IC50 between 100 nmol/L and 10 nmol/L (Fig. 5), even no inhibitory activity was observed against GIST48, GIST T1, and GIST882 [38, 39]. Ripretinib (6) got its first approval by the US FDA in May 2020 under the trade name QinlockTM for the treatment of adult patients with advanced gastrointestinal stromal tumors (GISTs) [40].

|
Download:
|
| Fig. 5. Structures of 6A. | |
{kind=link}
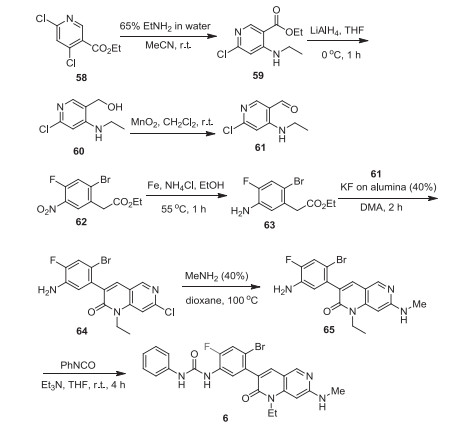
One synthetic method for the preparation of ripretinib (6) developed by Deciphera Pharmaceuticals is shown in Scheme 7 [38]. First, an aldehyde intermediate 61 was prepared, starting from ethyl 4, 6-dichloronicotinate (58). Amination substitution reaction of 4-chloro of ethyl 4, 6-dichloronicotinate (58) using 65% ethylamine in water at room temperature gave the intermediate 59, which was reduced by LiAlH4 at 0 ℃ to give alcohol 60. Oxidation of alcohol 60 with MnO2 at room temperature provided the aldehyde intermediate 61. On the other hand, the addition of iron powder and saturated ammonium chloride to ethyl 2-(2-bromo-4-fluoro-5-nitrophenyl)acetate (62) and heating at 55 ℃ for 1 h afforded amine 63. Then, the mixture of amine 63 and aldehyde intermediate 61 was sonicated in the presence of KF on alumina (40%) to give the cyclic compound 64. Next, compound 64 was treated with methylamine in a sealed tube and heated to 100 ℃. resulting in the amination compound 65. Finally, reaction of compound 65 with phenyl isocyanate in the presence of triethylamine at room temperature for 4 h to form the desired product 6 [38].

|
Download:
|
| Scheme 7. Synthesis of ripretinib (6). | |
{kind=link}
8. Rimegepant (Nurtec ODTTM)
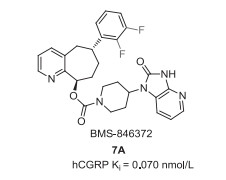
Rimegepant (7, BMS-927711) was discovered by Bristol-Myers Squibb in 2011, which was used as an oral calcitonin gene-related peptide (CGRP) antagonist for treating migraine in clinical trials. Rimegepant is a chiral compound featuring three chiral carbon centers and contains a cyclohepta[b]pyridine core, a bis-difluorinated phenyl moiety, and a β-alanine derived cyclic moiety (Fig. 1) [41, 42]. Previously, Bristol-Myers Squibb developed BMS-846372 (7A) as an oral CGRP receptor antagonist with a Ki value of 0.070 nmol/L (Fig. 6) [43]. Further SAR investigation based on the key difluorophenyl substituted cyclohepta[b]pyridine unit led to the discovery of rimegepant (7, BMS-927711) with the hCGRP Ki value of 0.027 nmol/L [41, 43]. In 2016, Biohaven Pharmaceutical entered into the development and commercialization of rimegepant. In February 2020, rimegepat received its first FDA approval for the acute treatment of migraine with or without aura in adults[44].

|
Download:
|
| Fig. 6. Structure of 7A. | |
{kind=link}
The synthetic route developed by Bristol-Myers Squibb for the preparation of rimegepant (7) is shown in Scheme 8 [41, 45]. Chiral alcohol 67 was generated via a Rh-catalyzed chemo- and enantioselective reduction of 7, 8-dihydro-5H-cyclohepta[b]pyridine-5, 9-dione (66) with 99% ee, which was protected by triisopropylsilyl triflate (TIPSOTf) to give compound 68 [46]. Then, compound 68 was subjected to the α-arylation reaction with 1-bromo-2, 3-difluorobenzene resulting in the ketone 69. Enantiomerically pure alcohol 70 could be easily obtained from dynamic resolution and diastereoselective reduction of ketone 69 with the use of Li(BuO)3AlH as a reductant in methyl tert-butyl ether (MTBE) [46]. Then, the treatment of alcohol 70 with NCS in the presence of triphenylphosphine in THF for 5 h affording chloride intermediate 71 with inversion of the chiral center. Substitution reaction of chloride intermediate 71 with sodium azide in DMF resulted in the azide 72 with the second inversion of the chiral center. Removal of triisopropylsilyl (TIPS) protecting group with TBAF in THF gave free alcohol 73, which was subjected to the substitution reaction to give the ester 75. The generated azide intermediate 75 was treated with trimethylphosphine in THF at room temperature for 2 h and followed by stirred with water for 3 h resulting in the desired rimegepant (7).

|
Download:
|
| Scheme 8. Synthesis of rimegepant (7). | |
{kind=link}
9. Fluoroestradiol F-18 (CeriannaTM)
Estrogen receptors (ER) are important prognostic biomarkers to visualize tumor progression/regression and targeted hormone treatment. The radioactive fluorine 18F molecules can accumulate in cancerous tissues expressing estrogen receptor, thus providing contrast agents for diagnosis. They can also be used for the assessment of heterogeneity in ER expression evading the requirement of biopsy [47]. A wide range of 18F radiopharmaceuticals used in PET has been reported in the literature [48]. The introduction of a small and highly electronegative fluorine atom as a hydrogen mimic may improve both pharmacokinetic and physicochemical properties, including binding affinity, metabolic stability and bioavailability. To develop high-affinity estrogen receptor-targeted diagnostic agents having the ability to cross the blood-brain barrier led to the synthesis of radioactive fluoroestradiols. These molecules have a high binding affinity for estrogen receptors and have high tissue permeability, including the blood-brain barrier [49].
Fluoroestradiol F-18 (8, also known as [18F]16α-fluoroestradiol) was developed by Zionexa USA and commercialized through Petnet Solutions, Zionexa USA's manufacture and exclusive distributor in the USA. The US FDA approved Fluoroestradiol F-18 on May 20, 2020, as a radioactive diagnostic agent for the non-invasive visual monitoring of ER-positive lesions during PET scan of patients with recurrent or metastatic breast cancer (Fig. 1). The agent provides enough contrast in PET scan images and helps diagnose multiple tumor sites without patient discomfort. The FDA approval was based primarily on the data from two published clinical trials [50, 51]. All the patients with recurrent or metastatic ER-positive breast cancer received radioactive fluoroestradiol before PET scan, and the results were compared to tissue biopsy data.
Kiesewetter et al. reported the first synthetic procedures for the preparation of 18F-labeled estrogens [52]. Other research groups [53] later reported the synthesis of fluoroestradiols. 18F fluorine required for synthesizing the fluoroestrogens was made from 18O-H2O by the 18O(p, n)18F reaction [52]. The 18F was eluted with a solution of 100 μL of 0.25 mol/L K2CO3 and 900 μL of K2.2.2 (15 mg/mL in MeCN) into the reaction vial. Compound 3-O-methoxymethyl-16, 17-O-sulphuryl-16-epiestriol (MMSE, 76) in anhydrous MeCN was added to azeotropically dried kryptofix-2.2.2 (K2.2.2)/K18F, and the mixture was heated at 110 ℃ for 15 min to provide intermediate 77. After 18F fluorination, the solution was quenched by heating the reaction mixture with 2.0 mol/L HCl (0.6 mL) for 10 min at 120 ℃ and then neutralized by adding 2.0 mL of 4.2% NaHCO3. The crude product 8 was purified by semi-preparative HPLC with 50% ethanol (Scheme 9) [54]. The radiochemical yield (decay corrected) based on re-solubilized 18F fluoride activity was 29-43%.

|
Download:
|
| Scheme 9. Radioactive synthesis of fluoroestradiol F–18 (8). | |
{kind=link}
10. Flortaucipir F-18 (TauvidTM)
Alzheimer's disease (AD) is a progressive neurodegenerative disorder wherein neurofibrillary tangles (NFTs) are formed due to hyperphosphorylation of microtubule stabilizing tau protein leading to its abnormal accumulation in the patients' brains [55-57]. Although no cure exists, early intervention is required to delay the symptoms with proper medication and disease management. Usually, the disorder is diagnosed based on the patient's medical history of memory loss and cognitive functions. Various tau-specific ligands have been reported in the literature[58].
Flortaucipir F-18 (9, also known as 18F-AV1451 or 18F-T807) was developed by Avid Radiopharmaceuticals, a wholly-owned subsidiary of Lilly. Approved by US FDA on May 28, 2020, flortaucipir F-18 is the first radioactive diagnostic agent to visually detect the density and differential distribution of these tau deposits in the brain of suspected patients using positron emission tomography (PET). Flortaucipir F-18 administered intravenously, crosses the blood-brain barrier, and interacts with the misfolded tau protein in the brain. It can be easily identified with a PET scan and helps diagnose the tau associated pathology (Fig. 1) [59].
In addition to a synthetic route reported by Holt's group [60], Frey, Scott and co-workers reported an updated route for fortaucipir F-18 (Scheme 10). A solution of N-Boc nitro-precursor of tauvid 78 in DMSO (0.5 mg in 500 μL) was added to a solution of dried 18F-fluoride (produced from 18F-fluoride with K2CO3 (3.5 mg in 500 µL of H2O) and kryptofix-2.2.2 (K2.2.2) (15 mg in 1 mL of EtOH). The mixture was heated to 130 ℃ for 10 min with constant stirring. After the completion of the reaction, the reaction mixture was cooled to 50 ℃, and purified by semi-preparative HPLC. The fluoro derivative 79 was eluted with 0.5 mL of EtOH followed by thermal deprotection to get flortaucipir F-18 (9) [61]. The radiochemical yield (non-corrected) of flortaucipir F-18 prepared using this method was 14% based on starting 18F-fluoride.

|
Download:
|
| Scheme 10. Radioactive synthesis of flortaucipir F-18 (9). | |
{kind=link}
11. Decitabine/cedazuridine (InqoviTM)
Inqovi is a fixed-dose combination of decitabine/cedazuridine developed by Astex Pharmaceuticals Inc., a subsidiary of Otsuka Pharmaceuticals. The US FDA approved it on July 7, 2020, for the treatment of myelodysplastic syndromes and chronic myelomonocytic leukaemia.
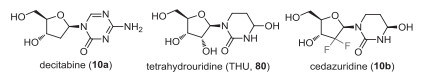
Decitabine (10a) is a hypomethylating nucleoside metabolic inhibitor, also known as cytidine antimetabolite, that after metabolism incorporates itself into DNA and inhibit DNA methyltransferase 1 (DNMT1) in proliferating cancer cells leading to re-activation of silenced tumor suppressor genes and apoptosis of cancerous cells restoring normal cellular differentiation and proliferation (Fig. 7) [62]. The oral bioavailability of this compound is limiting owing to its first-pass metabolism by the enzyme cytidine deaminase present in the gut, kidney and liver. Therefore, to facilitate its oral bioavailability and reduce high dose decitabine-associated gastrointestinal toxicity, its co-administration with cytidine deaminase (CDA) inhibitor-tetrahydrouridine (THU) (80) is suggested, and the patent was granted to Otsuka Pharmaceutical Co., Ltd. [63]. Poor aqueous solubility of THU and its isomerization at low pH reduce its potency in primates.

|
Download:
|
| Fig. 7. Structures of decitabine, cedazuridine, and tetrahydrouridine (THU). | |
{kind=link}
Both cytidine and deoxycytidine are being acted upon by CDA, proving that the 2′-hydroxy group of cytidine has no specific role in binding [64]. This has further been proved by the co-crystal structure of mouse CDA with THU, which shows that only the 3′-OH and 5′-OH groups of sugar form critical hydrogen bonds with the specific amino acid residues in the active site [65]. Later, 2′-deoxytetrahydrouridine was reported to be more potent than THU [66]. To improve the acid stability and increase the systemic exposure, Ferraris et al. described 2′-fluorinated tetrahydrouridine derivatives such as cedazuridine (10b) as new generation CDA inhibitors that prevent the breakdown and efficacy of decitabine in primates [67]. The acid-stable CDA inhibitor cedazuridine (10b) showed improved acid stability and better oral bioavailability in rhesus monkeys than tetrahydrouridine and its other analogs. Further, its co-administration with a CDA substrate decitabine showed increased plasma levels of decitabine in primates [67, 68]. The orientation of a 4-hydroxyl group of THU plays a critical role in the mechanism of inhibition of CDA, explaining the difference in potency of its two isomers. A similar binding mode/interaction with the active site zinc atom is observed for fluorinated analogs.
The US FDA approved Cedazuridine to combine with decitabine for sale by Astex Pharmaceuticals Inc. under the name Inqovi. The approval is based on phase Ⅲ studies that compared systemic exposure to decitabine from Inqovi and assessed its safety and efficacy [69].
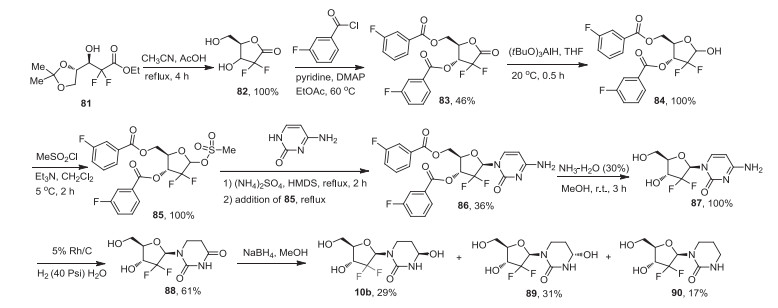
The fluorinated THU analogs were prepared using the procedures reported by the Sturla group for the synthesis of 2′-deoxytetrahydrouridine [70]. The synthesis of cedazuridine (10b) was published with the details of its development and pharmacological evaluation. As illustrated in Scheme 11, the key intermediate, gemcitabine (87) was prepared with chiral difluoro ester 81 as the starting reagent [70b]. Intramolecular cyclization reaction of compound 81 generated intermediate 82 in 100% yield, which was protected by 3-fluorobenzoyl group to give compound 83. Reduction of this lactone 83 by lithium tri-tert-butoxyaluminohydride at 20 ℃, followed by treatment with methanesulfonyl chloride in the presence of trimethylamine, resulting in the intermediate 85. Substitution reaction of intermediate 85 by cytosine in the presence of ammonium sulfate and 1, 1, 1, 3, 3, 3-hexamethyldisilazane (HMDS) afforded the intermediate 86, which was treated by 30% ammonia to give gemcitabine (87). Then, rhodium-catalyzed hydrogenation of compound 87 formed 2-deoxy-2, 2-difluorodihydrouridine (88). This was followed by the reduction of intermediate 88 with NaBH4 to give a mixture of difluorinated tetrahydrouridine epimers (10b) and (89) that differ in hydroxyl group stereochemistry at the position-4 along with 4-deoxy by-product 90. The preparative HPLC separated two epimers 10b and 89, and also removed the by-product 90.

|
Download:
|
| Scheme 11. Synthesis of cedazuridine (10b). | |
{kind=link}
12. Pralsetinib (BLU-667, GavretoTM)
First-generation kinase inhibitors for kinase-driven cancers like BCR-ABL fusions (imatinib), EGFR mutations (erlotinib and gefitinib), and ALK rearrangements (crizotinib) have been reported in the literature [71]. However, due to drug resistance development, search for next-generation kinase inhibitors with more potency and selectivity is desired [72]. Rearranged during transfection (RET) fusions have a receptor tyrosine kinase domain that is implicated in various cancers, including non-small cell lung cancer (NSCLC), medullary thyroid cancer (MTC), and papillary thyroid cancer [73]. In normal conditions, RET fusions are involved in kidney morphogenesis and embryonic development [74]. However, oncogenic RET kinase activation leads to tumorigenesis via point mutations and gene rearrangements [75, 76]. RET fusion proteins and specific activating point mutations lead to hyperactivation of downstream signaling cascades responsible for uncontrolled cell proliferation. The compounds like cabozantinib, vandetanib, sorafenib, regorafenib are active against the wild-type RET but are much less active against the mutated forms.
Subbiah et al. describe pralsetinib (11, also known as BLU-667) as an orally active, highly potent, and selective RET inhibitor for the treatment of metastatic RET fusion-positive non-small cell lung cancer (NSCLC) caused by abnormal RET (receptor tyrosine kinase rearranged during transfection) genes (Fig. 1) [77, 78]. More than 60 chemical scaffolds were tested and optimized to identify leads with improved RET potency and selectivity against other human kinases leading to the synthesis of pralsetinib. The efficacy of pralsetinib was evaluated in patients with RET fusion-positive metastatic NSCLC, and the compound showed increased RET selectivity and potency compared to multi-kinase inhibitors. The US FDA granted its approval to Blueprint Medicines on September 4, 2020.
The synthesis of pralsetinib was reported in a patent (Scheme 12) [79]. 2, 4-Dichloro-6-methylpyrimidine (91) was treated with MeSNa in THF to provide methylthiolated pyrimidine 92. Methyl 4-iodo-1-methoxycyclohexanecarboxylate (93) dissolved in dimethylacetamide is treated with Rieke Zinc and compound 92 followed by addition of PdCl2dppf to coupling product 94. Oxidation of sulfide in 94 with mCPBA afforded methylsulfonylpyrimidine 95, which was treated with acetic acid to afford 4-hydroxypyrimidine compound 96. Chlorination of 96 with POCl3 for 97 followed by chlorine-displacement with 3-methyl-1-pyrazol-5-amine (98) in the presence of Pd2(dba)3 and t-BuXPhos under heating gave compound 99. At the last step, the ester of compound 99 was hydrolyzed to acid, followed by benzotriazole-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP)-promoted amide coupling with compound 100 gave a mixture of diastereomers, and pralsetinib (11) was isolated by SFC as white solids.

|
Download:
|
| Scheme 12. Synthesis of pralsetinib (11). | |
{kind=link}
13. Berotralstat (OrladeyoTM)
Hereditary angioedema is a life-threatening rare genetic disorder caused by mutations in the regulatory regions of gene encoding a serine protease-C1 esterase inhibitor [80]. This inhibitor covalently binds and inactivates plasma kallikrein (PKal) - a protease responsible for cleaving a biologic peptide-kininogen, thereby preventing uncontrolled contact activation and release of a potent vasodilator, bradykinin. Due to over activation of the kallikrein–bradykinin cascade, either a deficiency (type Ⅰ) or dysfunction (type Ⅱ) of C1 esterase inhibitor is observed that leads to an uncontrolled increase of bradykinin, leading to increased vascular permeability and episodic swelling and pain [81].
The drugs available for oral prophylaxis against angioedema attacks are attenuated androgens, such as danazol and tranexamic acid. Due to various undesirable adverse androgenic hormonal effects and contraindications of these drugs, their clinical use is limited. Various groups have reported SAR studies of PKal inhibitors having different scaffolds like amino acid trimers, 1H-pyrazole-4-carboxamide/1H-imidazole-4-carboxamide, 3-(trifluoromethyl)-1H-pyrazole-5-carboxamide, 7H-purin-6-amine, nicotinamide, pyrrolidine-2-carboxamide, benzylamine. Many peptide-based PKal inhibitors having different scaffolds and selectivity have been developed, and a few of them have reached clinical trials or market [82]. Only two small-molecule non-peptidic pKal inhibitors were reported, BCX4161 (101) [83] and compound 102 (Fig. 8) [84]. BCX4161 failed in Phase 2 clinical trials due to its poor pharmacokinetics and efficacy. An orally bioavailable non-peptidic C1 inhibitor-BCX7353 (orladeyo) developed by BioCryst Pharmaceuticals Inc., is a potent oral small-molecule inhibitor of plasma kallikrein that performs its function by inhibiting its proteolytic activity and reducing bradykinin formation [85]. This inhibitor is the first oral non-steroidal medication approved by the US FDA on December 3, 2020, for the prophylactic treatment of hereditary angioedema that may help prevent angioedema attacks.

|
Download:
|
| Fig. 8. Structures of BCX4161 and compound 102. | |
{kind=link}
The synthetic route for berotralstat is shown in Scheme 13 [86]. The reaction of 3-formylbenzonitrile (103) and Grignard reagent 104 gave aniline derivative 105, which was used for PyBrO-promoted amide coupling with acid 106 to afford amide 107. The reaction of 107 with cyclopropylmethanamine in the presence of SOCl2 gave compound 108, which then converted to berotralstat (12) after de-Boc with HCl and chiral resolution.

|
Download:
|
| Scheme 13. Synthesis of berotralstat (12). | |
{kind=link}
14. Relugolix (OrgovyxTM)
Gonadotropin-releasing hormones (GnRH) are produced in the hypothalamus and released into the pituitary. These hormones interact with the GnRH receptor and activate the biosynthesis of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). The secretion of these hormones is controlled by feedback regulation from the hypothalamus [87].
GnRH antagonists bind competitively to GnRH receptors and decrease the release of LH and FSH from the anterior pituitary gland and, ultimately, decreases testosterone production in males and estrogen in females. Various peptidic GnRH antagonists, including GnRH-derived linear peptides [88], a cyclic hexapeptide derivative [89], a bicyclic peptide derivative [90] and so on, are reported in the literature. The problems associated with peptide-based compounds like metabolic stability and oral bioavailability created the strong need for a non-peptide based oral GnRH antagonist for sex-hormone-dependent cancers, e.g., prostatic cancer, endometriosis, etc.
To overcome this issue of poor metabolic stability and a short half-life of peptide-based drugs, orally active non-peptidic small molecules are required to treat these hormone-based reproductive disorders [91]. Various non-peptidic GnRH-antagonists [92] are reported to prevent or treat anterior pituitary hormone-dependent diseases. Abbott Laboratories reported the first non-peptide GnRH antagonist [93].
Orally active thienopyrimidine based selective GnRH receptor antagonist was first described in 2004 [94, 95]. Takeda Company developed a series of thienopyridine derivatives, including sufugolix and relugolix. Besides, various scaffolds of non-peptidic GnRH antagonists have been designed and tested [96].
The US FDA approved Relugolix (also known as TAK-385) in December 2020 as the first oral medication for treating advanced prostate cancer. The efficacy of relugolix was assessed by the percentage of participants who achieved and maintained a low testosterone level equal to castration [97]. It was the second orally active GnRH antagonist to be introduced for clinical use after elagolix (Orilissa) developed by Neurocrine Biosciences [98].
The bicyclic scaffold of thieno-[2, 3-d]pyrimidine-2, 4-dione core mimics a type Ⅱ β-turn of the human GnRH receptor. The structure-activity relationship (SAR) studies led to the identification of the compound, sufugolix showing good in vitro and in vivo GnRH antagonistic activities [99]. To improve in vivo GnRH antagonistic activity, Miwa et al. synthesized various analogs by performing modification at the 5-position of the thieno[2, 3-d]pyrimidine-2, 4-dione ring scaffold [95]. Further optimization was carried out by modification at 3- and 5-positions that resulted in relugolix (13) as a highly potent and orally active GnRH antagonist (Fig. 1). Optimized the thienopyrimidine ring at positions 5 and 3 by introducing polar substituents to increase GnRH antagonistic activity and reduction in CYP3A4 inhibition reported with earlier analogs. Using molecular modeling strategy, they observed that the 2, 6-difluorobenzyl group shows a hydrophobic interaction with Tyr283. The methoxyureido group interacts with both Gln106 and Asn102, and a tertiary amine forms a hydrogen bond with Asp302 of the GnRH receptor. Although substituting phenyl with pyridyl group leads to a decrease in logD, but CYP3A4 inhibition remained. The introduction of the hydroxymethyl group into the pyridyl ring and modifying the methylene linker could not reduce the CYP3A4 inhibitory activity. Further, no change was observed with the pyridyl ring's modification with the triazolyl ring or cyclic carboxamide group. But replacement with alkylsulfamoyl or methoxyethyl group leads to a decrease of logD value and the reduction of CYP3A4 inhibition. The SAR data showed that the methoxy group's introduction into the para position of a pyridine derivative leads to improved antagonistic activity with low CYP3A4 inhibitory activity. Further, to reduce the logD, the pyridine ring was replaced with a pyridazine ring. The derivative showed improved antagonistic activity with some CYP3A4 inhibition. To further reduce the logD, the N-methyl group was introduced in place of the N-2-methoxyethyl group. The dimethylaminomethyl pyridazine derivative showed potent antagonistic activities with no CYP3A4 inhibitory activity.
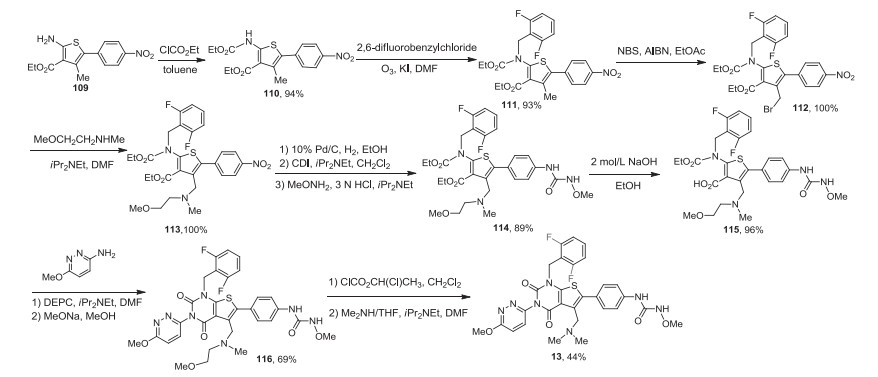
The synthesis of relugolix is shown in Scheme 14 [95]. 2-Aminothiophene derivative 109 was treated with ethoxycarbonychloride to form intermediate 110 followed by alkylation with 2, 6-difluorobenzylchloride to give N, N-disubstituted amine 111 which was brominated with N-bromosuccinimide (NBS) and 2, 2′-azobis(isobutyronitrile) (AIBN) to afford bromomethyl derivative 112. The reaction of 112 with N-(2-methoxyethyl)methylamine gave tertiary amine 113 followed by a one-pot reduction of NO2 to NH2 and the treatment with 1, 10-carbonyldiimidazole (CDI) and N-methoxyamine hydrochloride to give compound 114. Selective alkaline hydrolysis of ethyl ester yielded compound 115. The thiophene-3-carboxylic acid thus formed is condensed with various amines using diethyl phosphorocyanidate (DEPC) followed by cyclization under basic conditions using NaOEt provided the corresponding thieno[2, 3-d]pyrimidine-2, 4-dione derivatives 116. The reaction of 116 with 1-chloroethylchloroformate afforded the intermediary quaternary ammonium salts, and subsequent nucleophilic substitution with dimethylamine gave product relugolix (13).

|
Download:
|
| Scheme 14. Synthesis of relugolix (13). | |
{kind=link}
15. Conclusions and outlook
One of the striking trends observed for the drugs approved in 2020 is an overwhelming dominance of aromatic fluorination. While it is not completely unexpected, due to the advances in synthetic methodology, one would expect an increasing number of compounds featuring aliphatic fluorine. Nevertheless, some new fluorination motifs represented by KoselugoTM (3), TauvidTM (9) and PralsebinibTM (11), bearing fluorine on a heteroaromatic moiety. Also of interest are compounds PemazyreTM (4), Nurtec ODTTM (7), and OrgovyxTM (13), featuring two fluorine atoms on a phenyl ring. In this line, we also can mention QinlockTM (6) as an example of the fluorination-halogenation (Br) motif. Another example of multiple fluorinations is OrladeyoTM (12), possessing aromatic fluorine in combination with heteroaromatic trifluoromethyl group. Other cases of aromatic fluorination represented by AyvakitTM (1), IsturisaTM (2) and TabrectaTM (5) correspond to a rather established pattern of ortho- or para-substitution to increase the metabolic stability of a drug molecule. Examples of aliphatic fluorination are represented by CeriannaTM (8) (CHF) and InqoviTM (10) (CF2). In both cases, the fluorinated moiety is located next to a stereogenic center bearing hydroxy and amino functional groups, thus requiring an elaborated synthetic approach. As a manifestation of the growing importance of PET, functional imaging technique, is the approval of two new drugs CeriannaTM (8) and TauvidTM (9), bearing F18 nucleus, representing aliphatic and heteroaromatic fluorination, respectively. Another striking trend observed for the new pharmaceuticals in 2020 is that over 50% of compounds were developed as anticancer drugs, including AyvakitTM (1), PemazyreTM (4), TabrectaTM (5), QinlockTM (6), CeriannaTM (8), PralsebinibTM (11), OrgovyxTM (13). Other therapeutic areas represented by IsturisaTM (2), developed against Cushing's disease, KoselugoTM (3) to cure neurofibromatosis, Nurtec ODTTM (7) to treat migraine, TauvidTM (9) as a remedy for Alzheimer's disease, InqoviTM (10) to address myelodysplastic syndromes and PralsebinibTM (11) to cure hereditary angioedema (11). Similar to the previous years, over 50% of new drugs approved in 2020 are chiral compounds represented by AyvakitTM (1), IsturisaTM (2), Nurtec ODTTM (7), CeriannaTM (8), InqoviTM (10), PralsebinibTM (11) and OrladeyoTM (12). In this regard, we would like to stress the necessity of conducting the corresponding SDE-tests [25f] to ensure the accuracy of the reported values of the stereochemical outcome of catalytic enantioselective reactions. It is important to know that fluorine is an SDE-phoric substituent [6c], requiring a detailed study of the SDE properties of chiral fluorinated drugs to ensure their safe production storage and administration. Finally, we would like to point our recent concerns with fluoride overload in humans and the environment [3f]. Fluorine-containing drugs are considered as one of the sources of fluoride, calling for the necessity for more detailed and systematic studies of the metabolism of fluoro-organic products and the effect of fluoride on human health.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21761132021) and IKERBASQUE, Basque Foundation for Science.
| [1] |
(a) N. Aspern, S. Roser, B.R. Rad, et al., J. Fluorine Chem. 198 (2017) 24-33; (b) F. Gschwind, G. Rodriguez-Garcia, D.J.S. Sandbeck, et al., J. Fluorine Chem. 182 (2016) 76-90; (c) N. von Aspern, G.V. Roeschenthaler, M. Winter, I. Cekic-Laskovic, Angew. Chem. Int. Ed. 58 (2019) 15978-16000; (d) Y. Wang, W.H. Zhang, ChemElectroChem 2 (2015) 22-36; (e) T. Wang, X. Zang, X. Wang, et al., Energy Stor. Mater. 30 (2020) 367-384; (f) G.G. Amatucci, N. Pereira, J. Fluorine Chem. 128 (2007) 243-262; (g) W. Zhu, J.M. Alzola, T.J. Aldrich, et al., ACS Energy Lett. 4 (2019) 2695-2702; (h) T. Nakajima, J. Fluorine Chem. 105 (2000) 229-238; (i) Y. Che, X. Yuan, L. Sun, et al., J. Mater. Chem. C 8 (2020) 15839-15851; (j) X.B. Cheng, R. Zhang, C.Z. Zhao, et al., Adv. Sci. 3 (2016) 1500213. |
| [2] |
(a) T. Fujiwara, D. O'Hagan, J. Fluorine Chem. 167 (2014) 16-29; (b) M.G. Cagetti, G. Campus, E. Milia, P. Lingström, Acta Odontol. Scand. 71 (2013) 381-387; (c) Y. Lu, W.F. Guo, X.Q. Yang, J. Agric. Food Chem. 52 (2004) 4472-4476; (d) N.I. Sarker, M.S. Ahmad, S. Islam, N.H. Memon, Fluoride 53 (2020) 1-22; (e) L.A. Schaider, S.A. Balan, A. Blum, et al., Environ. Sci. Technol. Lett. 4 (2017) 105-111; (f) J. Cholak, J. Occup. Environ. Med. 1 (1959) 501-511; (g) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai, N. Shibata, iScience 23 (2020) 101467. |
| [3] |
(a) T. Fujiwara, D. O'Hagan, J. Fluorine Chem. 167 (2014) 16-29; (b) D. O'Hagan, J. Fluorine Chem. 131 (2010) 1071-1081; (c) W. Zhu, J. Wang, S. Wang, et al., J. Fluorine Chem. 167 (2014) 37-54; (d) J. Wang, M. Sanchez-Rosello, J.L. Acena, et al., Chem. Rev. 114 (2014) 2432-2506; (e) Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422-518; (f) J. Han, L. Kiss, H. Mei, et al., Chem. Rev. 121 (2021) 4678-4742; (g) M.A. Miller, E.M. Sletten, ChemBioChem 21 (2020) 3451-3462; (h) S.J. Fomon, J. Ekstrand, E.E. Ziegler, J. Public. Health Dent. 60 (2000) 131-139; (i) A. Strunecká, J. Patočka, P. Connett, J. Appl. Biomed. 2 (2004) 141-150; (j) D. Kanduti, P. Sterbenk, B. Artnik, Mater. Sociomed. 28 (2016) 133-137; (k) P.T. Lowe, D. O'Hagan, J. Fluorine Chem. 230 (2020) 109420. |
| [4] |
(a) L. Wang, T. Kitamura, Y. Zhou, et al., J. Fluorine Chem. 240 (2020) 109670; (b) J. Han, G. Butler, H. Moriwaki, et al., Molecules 25 (2020) 2116; (c) H. Mei, J. Han, S. White, G. Butler, V.A. Soloshonok, J. Fluorine Chem. 227 (2019) 109370; (d) 2019 A. Sato, M.V. Ponomarenko, T. Ono, G.V. Röschenthaler, V.A. Soloshonok, Eur. J. Org. Chem. (2019) 4417-4421. 2019; (e) W. Zhang, Chem. Rev. 104 (2004) 2531-2556; (f) W.B. Yi, J.J. Ma, L.Q. Jiang, C. Cai, W. Zhang, J. Fluorine Chem. 157 (2014) 84-105; (g) A.J. Cresswell, S.G. Davies, P.M. Roberts, J.E. Thomson, Chem. Rev. 115 (2015) 566-611; (h) J. Charpentier, N. Früh, A. Togni, Chem. Rev. 115 (2015) 650-682; (i) X. Liu, C. Xu, M. Wang, Q. Liu, Chem. Rev. 115 (2015) 683-730; (j) X.H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115 (2015) 731-764; (k) V.G. Nenajdenko, V.M. Muzalevskiy, A.V. Shastin, Chem. Rev. 115 (2015) 973-1050. |
| [5] |
(a) M.G. Campbell, T. Ritter, Chem. Rev. 115 (2015) 612-633; (b) L. Wang, Z. Li, J. Liu, et al., Curr. Org. Chem. 24 (2020) 2181-2191; (c) J. Liu, Z. Li, H. Mei, V.A. Soloshonok, J. Han, ACS Omega 4 (2019) 19505-19512; (d) H. Mei, J. Liu, S. Fustero, et al., Org. Biomol. Chem. 17 (2019) 762-775; (e) H. Mei, J. Han, S. Fustero, et al., J. Fluorine Chem. 216 (2018) 57-70; (f) Y. Zhu, J.L. Han, J. Wang, et al., Chem. Rev. 118 (2018) 3887-3964; (g) L. Zhang, W. Zhang, W. Sha, et al., J. Fluorine Chem. 198 (2017) 2-9; (h) J. Han, A.E. Sorochinsky, T. Ono, V.A. Soloshonok, Curr. Org. Synth. 8 (2011) 281-294; (i) J.L. Aceña, A.E. Sorochinsky, H. Moriwaki, T. Sato, V.A. Soloshonok, J. Fluorine Chem. 155 (2013) 21-38. |
| [6] |
(a) E. Tokunaga, H. Akiyama, V.A. Soloshonok, et al., PLoS ONE 12 (2017) e0182152; (b) A.E. Sorochinsky, T. Katagiri, T. Ono, et al., Chirality 25 (2013) 365-368; (c) A.E. Sorochinsky, J.L. Aceña, V.A. Soloshonok, Synthesis 45 (2013) 141-152; (d) D. O'Hagan, H. Deng, Chem. Rev. 115 (2015) 634-649; (e) I. Tirotta, V. Dichiarante, C. Pigliacelli, et al., Chem. Rev. 115 (2015) 1106-1129; (f) R. Hoque, A.L. Chesson, J. Clin. Sleep Med. 5 (2009) 471-476; (g) P. Venkateswarlu, Adv. Dent. Res. 8 (1994) 80-86; (h) X.G. Li, M. Haaparanta, O. Solin, J. Fluorine Chem. 143 (2012) 49-56; (i) M. Houde, J.W. Martin, R.J. Letcher, K.R. Solomon, D.C.G. Muir, Environ. Sci. Technol. 40 (2006) 3463-3473; (j) J. Yang, R. Zhang, Y. Zhao, et al., Spectrochim. Acta A 248 (2021) 119199. |
| [7] |
(a) H. Mei, J. Han, S. White, et al., Chem. Eur. J. 26 (2020) 11349-11390; (b) H. Mei, J. Han, K.D. Klika, et al., Eur. J. Med. Chem. 186 (2020) 111826; (c) J. Han, A.M. Remete, L.S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V.A. Soloshonok, D. O'Hagan, J. Fluorine Chem. 239 (2020) 109639; (d) K.L. Kirk, J. Fluorine Chem. 127 (2006) 1013-1029; (e) E.P. Gillis, K.J. Eastman, M.D. Hill, D.J. Donnelly, N.A. Meanwell, J. Med. Chem. 58 (2015) 8315-8359; (f) W.K. Hagmann, J. Med. Chem. 51 (2008) 4359-4369; (g) M. Inoue, Y. Sumii, N. Shibata, ACS Omega 5 (2020) 10633-10640; (h) R. Hevey, Chem. Eur. J. 27 (2021) 2240-2253; (i) M. Shevchuk, Q. Wang, R. Pajkert, et al., Adv. Synth. Catal. (2021), doi: 10.1002/adsc.202001464. |
| [8] |
H. Mei, J. Han, S. Fustero, et al., Chem. Eur. J. 25 (2019) 11797-11819. DOI:10.1002/chem.201901840 |
| [9] |
H. Mei, A.M. Remete, Y. Zou, et al., Chin. Chem. Lett. 31 (2020) 2401-2413. DOI:10.1016/j.cclet.2020.03.050 |
| [10] |
B.L. Hodous, J.L. Kim, K.J. Wilson, D. Wilson, WO2015057873 A1, 2015.
|
| [11] |
(a) E.K. Evans, A.K. Gardino, J.L. Kim, et al., Sci. Transl. Med. 9 (2017) 1690 eaao; (b) E.K. Evans, A. Gardino, B. Hodous, et al., Blood 126 (2015) 568. |
| [12] |
S. Dhillon, Drugs 80 (2020) 433-439. DOI:10.1007/s40265-020-01275-2 |
| [13] |
X. Xu, CN111454217 A, 2020.
|
| [14] |
R. Shintani, M. Takeda, T. Tsuji, T. Hayashi, J. Am. Chem. Soc. 132 (2010) 13168-13169. DOI:10.1021/ja106114q |
| [15] |
Y. Xu, Y. Yu, Z. Fan, L. Chen, CN110950872 A, 2020.
|
| [16] |
B.L. Hodous, US2016/0031892 A1, 2016.
|
| [17] |
K.L. Meredith, G. Ksander, L.G. Monovich, et al., ACS Med. Chem. Lett. 4 (2013) 1203-1207. DOI:10.1021/ml400324c |
| [18] |
J. Ménard, L. Pascoe, J. Hypertens. 24 (2006) 993-997. DOI:10.1097/01.hjh.0000226183.98439.b3 |
| [19] |
S. Duggan, Drugs 80 (2020) 495-500. DOI:10.1007/s40265-020-01277-0 |
| [20] |
C. Zhang, J. Chakma, WO2016/109361 A2, 2016.
|
| [21] |
(a) A.E. Sorochinsky, J.L. Aceña, V.A. Soloshonok, Synthesis 45 (2013) 141-152; (b) J. Han, O. Kitagawa, A. Wzorek, K.D. Klika, V.A. Soloshonok, Chem. Sci. 9 (2018) 1718-1739; (c) V.A. Soloshonok, D.O. Berbasov, Chim. Oggi. Chem. Today 24 (2006) 44-47. |
| [22] |
(a) A.E. Sorochinsky, T. Katagiri, T. Ono, et al., Chirality 25 (2013) 365-368; (b) T. Nakamura, K. Tateishi, S. Tsukagoshi, et al., Tetrahedron 68 (2012) 4013-4017; (c) Y. Suzukia, J. Han, O. Kitagawa, J.L. Aceñac, K.D. Klika, V.A. Soloshonok, RSC Adv. 5 (2015) 2988-2993. |
| [23] |
J.L. Aceña, A.E. Sorochinsky, T. Katagiri, V.A. Soloshonok, Chem. Commun. 49 (2013) 373-375. DOI:10.1039/C2CC37491A |
| [24] |
T. Katagiri, S. Takahashi, A. Tsuboi, M. Suzaki, K. Uneyama, J. Fluorine Chem. 131 (2010) 517-520. DOI:10.1016/j.jfluchem.2009.12.007 |
| [25] |
(a) H. Ueki, M. Yasumoto, V.A. Soloshonok, Tetrahedron 21 (2010) 1396-1400; (b) M. Yasumoto, H. Ueki, T. Ono, T. Katagiri, V.A. Soloshonok, J. Fluorine Chem. 131 (2010) 535-539; (c) M. Yasumoto, H. Ueki, V.A. Soloshonok, J. Fluorine Chem. 131 (2010) 266-269; (d) M. Yasumoto, H. Ueki, V.A. Soloshonok, J. Fluorine Chem. 131 (2010) 540-544; (e) J. Han, D.J. Nelson, A.E. Sorochinsky, V.A. Soloshonok, Curr. Org. Synth. 8 (2011) 310-317; (f) V.A. Soloshonok, A. Wzorek, K.D. Klika, Tetrahedron 28 (2017) 1430-1434. |
| [26] |
A. Markham, S.J. Keam, Drugs 80 (2020) 931-937. DOI:10.1007/s40265-020-01331-x |
| [27] |
E.L. Wallace, J.P. Lyssikatos, B.T. Hurley, A.L. Marlow, WO03077914, A1, 2003.
|
| [28] |
B.R. Davies, A. Logie, J.S. McKay, et al., Mol. Cancer Ther. 6 (2007) 2209-2219. DOI:10.1158/1535-7163.MCT-07-0231 |
| [29] |
C.J. Squire, J. Demattei, R.J. Boberts, WO2007076245 A2, 2007.
|
| [30] |
P.C.C. Liu, K. Holly, L. Wu, et al., PLoS ONE 15 (2020) e0231877. DOI:10.1371/journal.pone.0231877 |
| [31] |
L. Wu, C. Zhang, C. He, et al., WO2014007951 A2 (2014).
|
| [32] |
S.M. Hoy, Drugs 80 (2020) 923-929. DOI:10.1007/s40265-020-01330-y |
| [33] |
T.C. Burn, P.C. Liu, W. Frietze, et al., WO2019/213544 A2, 2019.
|
| [34] |
U. Glaenzel, Y. Jin, R. Hansen, et al., Drug Metab. Dispos. 48 (2020) 873-885. DOI:10.1124/dmd.119.090324 |
| [35] |
M. Xu, C. He, C. Zhang, WO2008/64157 A1, 2008.
|
| [36] |
J. Zhuo, C. He, W. Yao, US US2011/212967 A1, 2011.
|
| [37] |
S. Dhillon, Drugs 80 (2020) 1125-1131. DOI:10.1007/s40265-020-01347-3 |
| [38] |
D.L. Flynn, M.D. Kaufman, WO 2013/184119 A1, 2013.
|
| [39] |
D.L. Flynn, M.D. Kaufman, P.A. Petillo, US 8461179 B1, 2013.
|
| [40] |
S. Dhillon, Drugs 80 (2020) 1133-1138. DOI:10.1007/s40265-020-01348-2 |
| [41] |
G. Luo, L. Chen, C.M. Conway, et al., J. Med. Chem. 55 (2012) 10644-10651. DOI:10.1021/jm3013147 |
| [42] |
(a) J. Liu, J. Han, K. Izawa, et al., Eur. J. Med. Chem. 208 (2020) 112736; (b) Z. Yin, W. Hu, W. Zhang, et al., Amino Acids 52 (2020) 1227-1261. |
| [43] |
G. Luo, L. Chen, C.M. Conway, et al., ACS Med. Chem. Lett. 3 (2012) 337-341. DOI:10.1021/ml300021s |
| [44] |
L.J. Scott, Drugs 80 (2020) 741-746. DOI:10.1007/s40265-020-01301-3 |
| [45] |
G. Luo, GM. Bubowchik, J.E. Macor, WO 2011046997 A1, 2011.
|
| [46] |
D.K. Leahy, Y. Fan, L.V. Desai, et al., Org. Lett. 14 (2012) 4938-4941. DOI:10.1021/ol302262q |
| [47] |
(a) B.K. Park, N.R. Kitteringham, P.M. O'Neill, Annu. Rev. Pharmacol. Toxicol. 41 (2001) 443-470; (b) G.J. Liao, A.S. Clark, E.K. Schubert, D.A. Mankoff, J. Nucl. Med. 57 (2016) 1269-1275. |
| [48] |
(a) W. Li, Int. Congr. Ser. 1264 (2004) 53-76; (b) R.M. Glasspool, T.R.J. Evans, Eur. J. Cancer 36 (2000) 1661-1670; (c) L. Varagnolo, M.P.M. Stokkel, U. Mazzi, E.K.J. Pauwels, Nucl. Med. Biol. 27 (2000) 103-112; (d) M.J. Adam, D.S. Wilbur, Chem. Soc. Rev. 34 (2004) 153-163. |
| [49] |
(a) L.M. Peterson, D.A. Mankoff, T. Lawton, et al., J. Nucl. Med. 49 (2008) 367-374; (b) M. Paquette, É. Lavallée, S. Phoenix, et al., J. Nucl. Med. 59 (2018) 197-203. |
| [50] |
S.Y. Chae, S.H. Ahn, S.B. Kim, et al., Lancet Oncol 20 (2019) 546-555. DOI:10.1016/S1470-2045(18)30936-7 |
| [51] |
L.M. Peterson, B.F. Kurland, E.K. Schubert, et al., Mol. Imaging Biol. 16 (2014) 431-440. DOI:10.1007/s11307-013-0699-7 |
| [52] |
D.O. Kiesewetter, M.R. Kilbourn, S.W. Landvatter, et al., J. Nucl. Med. 25 (1984) 1212-1221. |
| [53] |
(a) P. Kumar, P.J. Mercer, C. Doerkson, K. Tonkin, A.J. McEwan, J. Pharm. Pharm. Sci. 10 (2007) 256s-265s; (b) J.L. Lim, L. Zheng, M.S. Berridge, T.J. Tewson, Nucl. Med. Biol. 23 (1996) 911-915. |
| [54] |
M. Dixit, J. Shi, L. Wei, G. Afari, S. Bhattacharyya, Int. J. Mol. Imaging (2013) 2786072013. |
| [55] |
M. Schöll, A. Maass, N. Mattsson, et al., Mol. Cell. Neurosci. 97 (2019) 18-33. DOI:10.1016/j.mcn.2018.12.001 |
| [56] |
V.L. Villemagne, M.T. Fodero-Tavoletti, C.L. Masters, C.C. Rowe, Lancet Neurol. 14 (2015) 114-124. DOI:10.1016/S1474-4422(14)70252-2 |
| [57] |
B. Hall, E. Mak, S. Cervenka, et al., Ageing Res. Rev. 36 (2017) 50-63. DOI:10.1016/j.arr.2017.03.002 |
| [58] |
A. Leuzy, K. Chiotis, L. Lemoine, et al., Mol. Psychiatry 24 (2019) 1112-1134. DOI:10.1038/s41380-018-0342-8 |
| [59] |
(a) G.J. Liao, A.S. Clark, E.K. Schubert, D.A. Mankoff, J. Nucl. Med. 57 (2016) 1269-1275; (b) Y.T. Wang, P. Edison, Curr. Neurol. Neurosci. Rep. 19 (2019) 45. |
| [60] |
D.P. Holt, H.T. Ravert, R.F. Dannals, J. Label. Compd. Radiopharm. 59 (2016) 411-415. DOI:10.1002/jlcr.3425 |
| [61] |
A.V. Mossine, A.F. Brooks, B.D. Henderson, et al., EJNMMI Radiopharm. Chem. 2 (2017) 7. DOI:10.1186/s41181-017-0027-7 |
| [62] |
(a) E. Jabbour, J.P. Issa, G. Garcia-Manero, H. Kantarjian, Cancer 112 (2008) 2341-2351; (b) K. Ueda, A. Masuda, M. Fukuda, et al., Drug Metab. Pharmacokinet. 32 (2017) 301-310; (c) M. Duchmann, R. Itzykson, Int. J. Hematol. 110 (2019) 161-169. |
| [63] |
S. Belyakov, G.S. Hamilton, B. Duvall, D. Ferraris, M. Vaal, AU2016213880B2, 2016.
|
| [64] |
(a) G.W. Camiener, Biochem. Pharmacol. 61 (1967) 1691-1702; (b) H. Hosono, S. Kuno, J. Biochem. 74 (1973) 797-803. |
| [65] |
A.H. Teh, M. Kimura, M. Yamamoto, et al., Biochemistry 45 (2006) 7825-7833. DOI:10.1021/bi060345f |
| [66] |
S. Costanzi, S. Vilar, D. Micozzi, et al., ChemMedChem 6 (2011) 1452-1458. DOI:10.1002/cmdc.201100139 |
| [67] |
D. Ferraris, B. Duvall, G. Delahanty, et al., J. Med. Chem. 57 (2014) 2582-2588. DOI:10.1021/jm401856k |
| [68] |
(a) A. Oganesian, S. Redkar, P. Taverna, G. Choy, R. Joshi-Hangal, M. Azab, Blood 122 (2013) 2526; (b) M.R. Savona, O. Odenike, P.C. Amrein, et al., Lancet Haematol. 6 (2019) e194-e203; (c) G. Garcia-Manero, E.A. Griffiths, D.P. Steensma, et al., Blood 136 (2020) 674-683. |
| [69] |
S. Dhillon, S. Drugs 80 (2020) 1373-1378. DOI:10.1007/s40265-020-01389-7 |
| [70] |
(a) J. Norton, H. Matsuo, S.J. Sturla, J. Org. Chem. 74 (2009) 2221-2223; (b) M.S. Kim, Y.J. Kim, J.H. Choi, H.G. Lim, D.W. Cha, WO2007/069838A1 (2007). |
| [71] |
(a) Jr G.Y. Ku, B.A. Haaland, G. de Lima Lopes, Lung Cancer 74 (2011) 469-473; (b) S.G. O'Brien, F. Guilhot, R.A. Larson, et al., New Engl. J. Med. 348 (2003) 994-1004; (c) A.T. Shaw, D.W. Kim, K. Nakagawa, et al., New Engl. J. Med. 368 (2013) 2385-2394; (d) C. Zhou, Y.L. Wu, G. Chen, et al., Lancet Oncol. 12 (2011) 735-742. |
| [72] |
(a) J.C. Soria, Y. Ohe, J. Vansteenkiste, et al., New Engl. J. Med. 378 (2018) 113-125; (b) J.J. Lin, A.T. Shaw, Trends Cancer 2 (2016) 350-364; (c) J.F. Gaino, A.T. Shaw, J. Clin. Oncol. 31 (2013) 3987-3996; (d) S. Peters, D.R. Camidg, A.T. Shaw, et al., New Engl. J. Med. 377 (2017) 829-838; (e) E. Jabbour, H. Kantarjian, J. Cortes Clin, Lymphoma Myeloma Leuk 15 (2015) 323-334. |
| [73] |
(a) J.F. Gainor, J.F. Shaw, Genome. Res. 3 (2013) 604-606; (b) Y.S. Ju, W.C. Lee, J.Y. Shin, et al., Genome. Res. 22 (2012) 436-445; (c) T. Kohno, H. Ichikawa, Y. Totoki, et al., Nat. Med. 18 (2012) 375-377; (d) N. Stransky, E. Cerami, S. Schalm, J.L. Kim, C. Lengauer, Nat. Commun. 5 (2014) 4846-4856; (e) P. Ballerini, S. Struski, C. Cresson, et al., Leukemia 26 (2012) 2384-2389; (f) A.F. Le Rolle, S.J. Klempner, C.R. Garrett, et al., Oncotarget 6 (2015) 28929-28937; (g) A. Skalova, G. Stenman, R.H.W. Simpson, et al., Am. J. Surg. Pathol. 42 (2018) e11-e27. |
| [74] |
M. Takahashi, J. Ritz, G.M. Cooper, Cell 42 (1985) 581-588. DOI:10.1016/0092-8674(85)90115-1 |
| [75] |
S.M. Jhiang, Oncogene 19 (2000) 5590-5597. DOI:10.1038/sj.onc.1203857 |
| [76] |
A. Russo, A.R. Lopes, M.G. McCusker, et al., Curr. Oncol. Rep. 22 (2020) 48. DOI:10.1007/s11912-020-00909-8 |
| [77] |
V. Subbiah, J.F. Gainor, R. Rahal, et al., Cancer Discov. 8 (2018) 836-849. DOI:10.1158/2159-8290.CD-18-0338 |
| [78] |
V. Subbiah, D. Yang, V. Velcheti, A. Drilon, F. Meric-Bernstam, J. Clin. Oncol. 38 (2020) 1209-1221. DOI:10.1200/JCO.19.02551 |
| [79] |
J.D. Brubaker, J.L. Kim, K.J. Wilson, D. Wilson, L.V. Dipietro, WO2017079140, 2017.
|
| [80] |
J.H. Levy, D.J. Freiberger, J. Roback, Anesth. Analg. 110 (2010) 1271-1280. DOI:10.1213/ANE.0b013e3181d7ac98 |
| [81] |
(a) J.W. Bryant, Z. Shariat-Madar, Cardiovasc. Hematol. Agents Med. Chem. 3(3) (2009) 234-250; (b) A.P. Kaplan, World Allergy Organ J. 4 (2011) 73-75; (c) A.H. Schmaier, Front. Med. (Lausanne) 5 (2018) 3. |
| [82] |
(a) Z. Xie, Z. Li, Y. Shao, C. Liao, Eur. J. Med. Chem. 190 (2020) 112137; (b) Z. Li, J. Partridge, A. Silva-Garcia, et al., ACS Med. Chem. Lett. 8 (2016) 185-190. |
| [83] |
E. Aygören-Pürsün, M. Magerl, J. Graff, et al., J. Allergy Clin. Immunol. 138 (2016) e5934-936. |
| [84] |
T. Brandl, S. Flohr, S. Kopec, et al., WO2012017020A1 (2012).
|
| [85] |
(a) J.R. Hwang, G. Hwang, A. Johri, T. Craig, Immunotherapy 17 (2019) 1439-1444; (b) E. Aygören-Pürsün, A. Bygum, V. Grivcheva-Panovska, et al., N. Engl. J Med. 379 (2018) 352-362. |
| [86] |
Y. El-Kattan, Y.S. Babu, WO2020092898A1, 2019.
|
| [87] |
N. Das, T.R. Kumar, J. Mol. Endocrinol. 60 (2018) R131-R155. DOI:10.1530/JME-17-0308 |
| [88] |
(a) T.D. Fitapatrick, J. Green, F. Haviv, C.A. Palabrica, US5140009, 1992; (b) T. Janaky, A. Juhasz, A.V. Schally, US5171835A, 1992. |
| [89] |
K. Kato, Y. Sugiura, K. Kato, US5633248, 1997.
|
| [90] |
R.J. Bienstock, J. Rizo, S.C. Koerber, et al., J. Med. Chem. 36 (1993) 3265-3273. DOI:10.1021/jm00074a006 |
| [91] |
(a) P. Limonta, M. Montagnani M. Marelli, S. Mai, et al., Endocr. Rev. 33 (2012) 784-811; (b) P. Limonta, M. Manea, Cancer Treat. Rev. 39 (2013) 647-663. |
| [92] |
(a) S. Furuya, N. Choh, K. Kato, S. Hinuma, WO95/28405, 1995; (b) S. Furuya, N. Choh, K. Kato, S. Hinuma, WO96/24597, 1996; (c) S. Furuya, N. Choh, S. Sasaki, WO97/14682, 1997; (d) S. Furuya, N. Choh, M. Harada, S. Sasaki, WO97/14697, 1997; (e) S. Furuya, T. Imaeda, S. Sasaki, WO99/33831, 1999; (f) S. Furuya, N. Choh, N. Suzuki, T. Imada, WO00/00493, 2000; (g) S. Furuya, N. Suzuki, N. Choh, Y. Nara, WO00/56739, 2000. |
| [93] |
B. De, J.J. Plattner, E.N. Bush, et al., J. Med. Chem. 32 (1989) 2036-2038. DOI:10.1021/jm00129a003 |
| [94] |
N. Cho, T. Imada, T. Hitaka, et al., US7300935B2, 2007.
|
| [95] |
K. Miwa, T. Hitaka, T. Imada, et al., J. Med. Chem. 54 (2011) 4998-5012. DOI:10.1021/jm200216q |
| [96] |
F.L. Tukun, D.E. Olberg, P.J. Riss, I. Haraldsen, A. Kaass, J. Klaveness, Mol. 22 (2017) 2188. DOI:10.3390/molecules22122188 |
| [97] |
A. Markham, Drugs 79 (2019) 675-679. DOI:10.1007/s40265-019-01105-0 |
| [98] |
(a) Y.N. Lamb, Drugs 78 (2018) 1501-1508; (b) "Elagolix-Abbvie/Neurocrine Biosciences-AdisInsight" (https://adisinsight.springer.com/drugs/800020238). |
| [99] |
T. Hara, H. Araki, M. Kusaka, et al., J. Clin. Endocrinol. Metab. 88 (2003) 1697-1704. DOI:10.1210/jc.2002-021065 |