 2021, Vol. 32
2021, Vol. 32 


b Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China;
c Nanjing IPE Institute of Green Manufacturing Industry, Nanjing 211100, China;
d School of Chemistry and Materials Science, Jiangsu Key Laboratory of New Power Batteries, Nanjing Normal University, Nanjing 210023, China
After significant successes in large amount of nanomaterials with homogeneous structures, e.g., sizes and morphologies [1-10], the research interest has turned to single or multicomponent based heterogeneous nanostructures, which are defined as a hybrid structure consisting of two or more nanoscale domains with distinct differences in chemical compositions or physical characteristics [11-21]. The attraction of these heterogeneous nanostructures roots in their possible synergetic effect due to the cooperation/interaction among different components or nanoscale domains, which is found to be prevail in many nanomaterials with heterogeneous structures, and often leads to much higher performance or improved property for a given application than the arithmetic sum of individual components [22-25]. As a typical example, owing to their localized surface plasmon resonance (LSPR) interaction between Au and copper selenide domains, the heterogeneous Au–Cu2−xSe nanocomposites exhibit two types of LSPR, but their features are apparently different from that of pure Au and that of bare Cu2−xSe nanoparticles [26].
A number of strategies based on wet-chemistry methods have been proposed and proven to be applicable for the synthesis of heterogeneous nanostructures with desired properties, among which the seed-mediated growth, galvanic replacement, and ionic exchange methods are capable to produce nanomaterials with heterogeneous structures including core-shell, cage-bell, dendritic, stellated, hollow, and composite/hybrid [27-38]. There have been many nice reviews to summarize the synthesis, properties, and applications of metal, semiconductor, and metal oxide-based heterogeneous nanostructures [23, 39-45]. Therefore, we limit the content of this review to the heterogeneous nanostructures derived from a special platform, i.e., Ag–Pt nanoparticles with core-shell constructions. The advantages of using core-shell Ag–Pt nanoparticles as starting materials to synthesize other heterogeneous nanostructures could be summarized as below: (1) A lot of reliable strategies for the synthesis of core-shell Ag–Pt nanoparticles have been developed and well documented; (2) more importantly, there is a unique phenomenon in core-shell Ag–Pt nanoparticles, i.e., the inside-out diffusion of Ag in core-shell Ag–Pt nanoparticles, due to the polyhedral morphologies of Ag seeds, which can be used to prepare a large amount of heterogeneous nanostructures; (3) Pt is the most important noble metal in electrochemical reactions such as methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR), and the Pt-based heterogeneous nanostructures derived from core-shell Ag–Pt nanoparticles have many welcome features (e.g., electronic interaction and unique morphologies) for catalyzing these electrochemical reactions. Therefore, in this review, we highlight the effective strategies developed for producing heterogeneous nanostructures using core-shell Ag–Pt nanoparticles as starting materials, including hollow, dimeric and composite structures as well as their related electrocatalytic applications. In the final section of this review, we also make an expectation for more heterogeneous nanostructures that may be prepared by using this core-shell precursor so as to offer the readers the opportunities and challenges in this specific realm.
2. Synthesis of core-shell Ag–Pt nanoparticlesIn principle, core-shell Ag–Pt nanoparticles can be prepared in aqueous phase by reducing Pt2+/Pt4+ ions in the presence of previously formed Ag seeds. Unfortunately, the galvanic replacement reaction between Ag seeds and Pt2+/Pt4+ ions in aqueous solution would interfere with the growth of Pt on Ag seeds, resulting in alloy Ag/Pt nanoparticles with hollow interiors instead of core-shell structures [46-61]. To avoid the replacement reaction between Ag seeds and Pt2+/Pt4+ ions, Aslam and Linic firstly modified the Pt2+ ions with OH− through ligand exchange for reducing their reduction potential. They then reduced the Pt2+ ions by ascorbic acid in the presence of Ag cubic seeds to obtain Ag–Pt core-shell nanocubes with ultrathin (< 1 nm) Pt layers [62]. However, these Ag–Pt core-shell particles were not used as starting materials to prepare other heterogeneous nanostructures. Instead, the core-shell Ag–Pt precursors used to derive heterogeneous nanostructures could be obtained with high yield and quality in an organic medium by seed-mediated growth at an appropriate temperature [63-65]. In brief, Ag seed particles were firstly prepared by reducing AgNO3 in oleylamine at 150 ℃, followed by the introduction of Pt2+ ions (K2PtCl4). The mixture was then heated at 160 ℃ for 1 h for completing the growth of Pt shell on the surfaces of Ag seeds. The as-prepared core-shell Ag–Pt nanoparticles could be readily collected from oleylamine by using methanol precipitation and re-dispersed in toluene. Figs. 1a and b show the transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of the core-shell Ag–Pt nanoparticles prepared by this way. They are uniform with an average diameter of ca. 14 nm, and their core-shell construction could be verified by elemental profiles of Ag and Pt recorded under scanning TEM (STEM) mode [63].

|
Download:
|
| Fig. 1. TEM (a, c) and HRTEM images (b, d) of core-shell Ag–Pt nanoparticles as-prepared in oleylamine at elevated temperature by seed-mediated growth method (a, b) and one-pot approach (c, d), respectively. Copied with permission [63]. Copyright 2012, the American Chemical Society; Copied with permission [66]. Copyright 2015, Royal Society of Chemistry. | |
{kind=link}
Further, the synthesis of core-shell Ag–Pt nanoparticles can be simplified to a one-step approach due to the difference in reduction kinetics of Ag+ and Pt2+ ions in oleylamine, whereby Ag nanoparticles are firstly formed for higher reduction potential of Ag+ ions, and then serve as seeds for Pt to nucleate and grow, resulting in a one-pot formation of Ag–Pt nanoparticles with core-shell structures [66]. Figs. 1c and d are the TEM and HRTEM images of the core-shell Ag–Pt nanoparticles obtained by the one-pot synthesis, which do not show apparent difference from those of core-shell Ag–Pt nanoparticles prepared by the seed-mediated growth. It is noteworthy that the reduction of sole Pt2+ ions in oleylamine under same experimental conditions only results in the formation of Pt nanoparticles with worm-like morphologies [67, 68].
3. Diffusion of Ag in core-shell Ag–Pt nanoparticles from inside to outsideAs has been revealed, the structure of core-shell Ag–Pt nanoparticles is not stable whether in colloidal or solid state, and the Ag in the core region could diffuse from inside to outside in core-shell Ag–Pt nanoparticles after aging them in colloidal solution or in solid state for a period of time [63, 64]. As shown by Figs. 2a and b for the TEM and HRTEM images of core-shell Ag–Pt nanoparticles after aging in toluene for 7 months, the original core-shell particles become a mixture composed of isolated Ag hollow structured Pt nanoparticles.

|
Download:
|
| Fig. 2. TEM image (a) and HRTEM image (b) of core-shell Ag–Pt nanoparticles after aging them in toluene for months at room temperature. (c) Schematic illustration showing the mechanism of Ag diffusion in core-shell Ag–Pt nanoparticles from inside to outside. Copied with permission [63]. Copyright 2012, the American Chemical Society. | |
{kind=link}
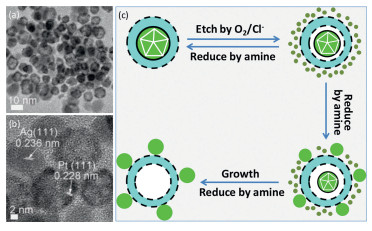
The diffusion of Ag in core-shell Ag–Pt nanoparticles is closely associated with the multiply twinned structure of Ag seeds and can be depicted by a mechanism shown in Fig. 2c [63]. For Ag seeds, their twined structure is not stable in nature, and can be slowly etched by O2 dissolved in the solution [5, 69]. In addition, the twin defects in Ag seeds would also strongly affect quality of the Pt shells formed on Ag seeds, e.g., roughness and discontinuity. Thus, as schematically indicated by Fig. 2c, the Ag cores are firstly etched into Ag+ ions by dissolved O2, which then diffuse from inside to outside through the discontinuous Pt shells due to the concentration gradient between internal and external regions of the core-shell particles. The diffused Ag+ ions could be reduced back into Ag particles with a single crystalline phase, which are stable to resist the O2 etching. Eventually, the original core-shell Ag–Pt nanoparticles are replaced by a physical mixture consisting of Pt nanoparticles with hollow interiors and Ag nanoparticles with single crystallinities. The inside-out diffusion of Ag is not limited in core-shell Ag–Pt nanoparticles, and can also proceed in core-shell structures with other monometallic metal shells (e.g., Ru, Ir, Rh, and Os) or alloy shells (e.g., PtRu, PtRh, PtOs, PtIr, and PtRuOs) [63]. This interesting diffusion phenomenon from inside to outside is a prerequisite for deriving heterogeneous nanostructures using core-shell Ag–Pt nanoparticles as starting materials, as we will review in the following sections.
4. Pt nanoparticles with hollow interiorsMaking Pt nanoparticles with hollow interiors is the most common practice to enhance their catalytic performance for a given application. Hollow structured Pt nanomaterials have two significant advantages: (i) The hollow interiors can save a lot of valuable Pt metal; (ii) the internal area could participate in the catalytic reactions pending that Pt shells are porous to allow the reactants/products to enter and exit. For example, hollow structured Pt nanoparticles with tiny channels in their shells are twice as active as their solid counterparts with analogous sizes and morphologies for MOR, the anodic reaction in direct methanol fuel cells (DMFCs) [70, 71]. In comparison with the abundant sacrificial templates used to prepare hollow structured metal oxide nanoparticles, e.g., polymer and inorganic spheres [72-78], liquid and microemulsion droplets [79-82], and vesicles [83-85], the templates used to derive Pt nanoparticles with hollow interiors are still limited to several metal seeds with relatively high reduction potentials, e.g., Ag and Co [70, 86, 87]. Typically, as shown in Fig. 3 for the overall scheme, Chen et al. reported a three-step strategy to synthesize hollow Pt spheres with nano-channels [86]. In the first step uniform Ag nanoparticles were synthesized, followed by a galvanic replacement reaction with an aqueous solution of H2PtCl6 at room temperature (second step), which transforms the Ag nanoparticles into alloy Ag/Pt shells. The simultaneously formed AgCl roughens the surface of the alloy shells, which is favorable for creating higher surface areas for catalytic reactions. In the last step (third step), Ag and AgCl in the alloy Ag/Pt nanoshells were removed by aqueous ammonia and HNO3 solution, producing the final hollow Pt products with nano-channels in their shells.

|
Download:
|
| Fig. 3. Schematic illustration showing the synthetic route for porous hollow Pt nanospheres with nano-channels. Copied with permission [86]. Copyright 2008, the American Chemical Society. | |
{kind=link}
Based on the outward diffusion of Ag in core-shell Ag–Pt nanoparticles, the core-shell Ag–Pt nanoparticles synthesized in organic medium could be used as starting materials for conveniently generating hollow structured Pt nanoparticles with uniform size distributions [63]. The completion of Ag inside-out diffusion in core-shell Ag–Pt nanoparticles usually takes months. However, this diffusion process can be greatly accelerated by a bis(p-sulfonatophenyl)phenylphosphane dihydrate dipotassium salt (BSPP), a chemical agent that binds strongly with Ag+ ions to form water-soluble coordinating compounds [71, 88]. BSPP therefore drives the inside-out diffusion of Ag in core-shell Ag–Pt nanoparticles, enabling its completion in 24−28 h, and leaving behind an organosol of hollow structured Pt nanoparticles, as shown by their TEM and HRTEM images (Figs. 4a and b), in which the hollow interiors could be easily identified by the image contrast between the middle and border regions.

|
Download:
|
| Fig. 4. TEM (a) and HRTEM image (b) of hollow structured Pt nanoparticles. Copied with permission [63]. Copyright 2012, the American Chemical Society. TEM images of carbon-supported hollow Pt nanoparticles (c, d) obtained by Na2S (c) or NaCl treatment (d). Insets are the corresponding HRTEM images of a single particle. Copied with permission [89]. Copyright 2014, the American Chemical Society. | |
{kind=link}
BSPP is too expensive to be largely used to produce Pt nanoparticles with hollow interiors. Thus, Liu et al. developed a cost-effective approach to prepare hollow Pt nanostructures supported on carbon substrates [89]. Their strategy involves the synthesis of core-shell Ag–Pt nanoparticles, the loading on carbon substrates and the removal of Ag cores by saturated Na2S or NaCl solution. After loading on carbon substrates but before the Na2S or NaCl treatment, the core-shell Ag–Pt nanoparticles were subjected to refluxing in acidic acid to protonate and remove the oleylamine surfactant capped on the particle surfaces. Agitating the core-shell Ag–Pt nanoparticles on carbon substrates with Na2S or NaCl would firstly result in the formation of Ag2S or AgCl precipitates, which are subsequently dissolved by saturated Na2S or NaCl solution through a reaction in term of Ag2S + Na2S → 2AgNaS2 or AgCl + NaCl → AgNaCl2, leading to the final formation of hollow structured Pt nanoparticles, as shown by Figs. 4c and d for the corresponding TEM images. It is noteworthy that the hollow structured Pt nanoparticles exhibit different electrocatalytic activity for MOR although both of them have comparable average sizes. In brief, the carbon-supported hollow Pt nanostructures obtained by NaCl treatment have higher electrocatalytic activity for MOR than that of hollow products obtained by Na2S treatment, probably because S2− anions can easily poison the Pt particle surfaces, inducing serious decrease of the surface areas for the catalytic methanol oxidation [89].
5. Pt nanoparticles with cage-bell structuresUpon the advantages of hollow and core-shell particles, the continuing research interests naturally turned to create hybrid nanostructures composed of core-shell and hollow structures, or a core-shell structure with a core-void-shell configuration, which are often named as cage-bell, yolk-shell or rattle-type structures. Combining the properties of the movable core with the shell with controlled physical features, e.g., porosity and composition, the cage-bell structured (CBS) nanoparticles have displayed huge application potentials in batteries [90-94], nanoreactors [95-97], drug delivery [98-100], magnetics [101], photonics [102], optical sensing [103] and photocatalysis [104].
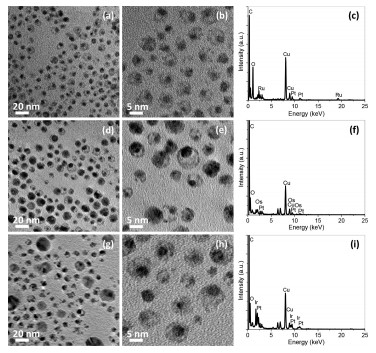
With slight modifications, the protocols for preparing hollow structured Pt nanoparticles can be used to direct the CBS nanoparticles with a movable Pt core and different metal shells [63, 105]. In this strategy, core-shell-shell Pt-Ag-M nanoparticles were firstly fabricated as starting templates, in which M refers the different metals at shell and Ag is placed at the inner shell region. The core-shell-shell Pt-Ag-M templates were then treated by aqueous BSPP solution or saturated NaCl aqueous solution to remove the inner Ag shell, resulting in the formation of CBS Pt-M nanoparticles in organic phase. Fig. 5 shows the CBS Pt-Ru, CBS Pt-Os and CBS Pt-Ir nanoparticles prepared by this way, in which the voids between the Pt core and the outer Ru, Os, Or Ir shell, formed due to the removal of inner Ag shell by saturated NaCl solution, could be clearly observable by brightness contrasts in their TEM images. In addition, the corresponding energy dispersive X-ray (EDX) spectra (Figs. 5c, f, and i) show the retaining of core and shell metals in the final CBS products, while the Ag disappearance directly proves the successful elimination of inner Ag shells after the NaCl treatment.

|
Download:
|
| Fig. 5. TEM images (a, d, g), HRTEM images (b, e, h), and corresponding EDX spectra (c, f, i) of the as-prepared CBS Pt-Ru (a, b, c), Pt-Os (d, e, f), and Pt-Ir nanoparticles (g, h, i). Copied with permission [105]. Copyright 2015, the Springer-Nature. | |
{kind=link}
CBS nanoparticles with Pt core and different metal shells have a unique application as electrocatalysts for oxygen reduction reaction, the cathodic reactions of direct methanol fuel cells (DMFCs). As verified by Feng et al., the CBS Pt-M nanoparticles exhibit comparable activity with that of the sole Pt particles with same sizes, as shown by Fig. 6a for the ORR polarization curves over various catalysts, meaning that the porous Ru, Os, or Ir shell in CBS particles does not have apparent effect on the ORR activity of Pt. However, they also found that the activities of CBS Pt-M nanoparticles for MOR are quite poor, also much lower than that of sole Pt particles (Fig. 6b). The different catalytic behavior of CBS Pt-M nanoparticles for ORR and MOR is believed to associate with the porous Ru, Os, or Ir shell, which prevents the methanol from contacting the Pt cores of CBS particles. Fortunately, although shielded by porous metal shells, the Pt cores in CBS particles are still accessible for oxygen molecules due to their smaller diameter (0.34 nm) compared with that of methanol (0.44 nm) [105].

|
Download:
|
| Fig. 6. ORR polarization curves for Pt/C, CBS Pt-Ru/C, CBS Pt-Os/C, and CBS Pt-Ir/C catalysts in an O2-saturated HClO4 solution (0.1 mol/L) at 20 mV/s and a rotating speed of 1600 rpm (a) and cyclic voltammograms of Pt/C, CBS Pt-Ru/C, CBS Pt-Os/C, and CBS Pt-Ir/C catalysts in argon-purged HClO4 (0.1 mol/L) with 1 mol/L methanol at 20 mV/s (b). Copied with permission [105]. Copyright 2015, the Springer-Nature. | |
{kind=link}
This selective activity for ORR rather than MOR of CBS Pt-M nanoparticles is of very importance, and it offers a concept to overcome the methanol crossover, one of the major obstacles for the commercialization of DMFCs [106-110], through a geometric design instead of making use of the intrinsic properties of the catalytic metals, which may also shed some light for developing other catalysts with strict requirements in selectivity. By elaborately tailoring the size of the inner core and the porosity of the metal shells in CBS particles, we can expect further enhancement of their performances, i.e., the catalytic activity selectivity, for oxygen reduction reaction at the cathode electrode of DMFCs.
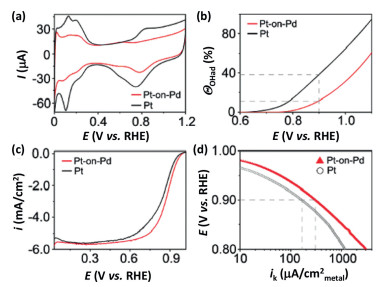
6. Pt nanoparticles with stellated morphologiesWith the combination of synergistic effect between different component domains and abundant steps, edges, corner atoms in branched shells, core-shell nanoparticles with stellated or dendritic shells have attracted extensive research interests [11, 106-113]. These heterogeneous core-shell nanostructures, also called particle-on-particle structures, usually have enhanced catalytic properties in comparison with that of each one of the constituent components, and this could be demonstrated by the electrocatalytic oxygen reduction over heterogeneous Pt-on-Pd bimetallic nanostructures [114-117]. Through a facet-selective growth, a dense array of Pt dendrites can be formed on the Pd seeds with truncated shapes, and these Pt-on-Pd bimetallic nanostructures are found to be 2.5 times more active than the commercial Pt/C catalyst for ORR, as evinced by Fig. 7 for the electrochemical measurements [115].

|
Download:
|
| Fig. 7. Cyclic voltammograms (a), hydroxyl surface coverage (ΘOH) (b), ORR polarization curves (c), and specific kinetic current densities (ik) for carbonsupported Pt-on-Pd and Pt catalysts (d). Copied with permission [115]. Copyright 2009, the American Chemical Society. | |
{kind=link}
Kim et al. offered another example to use the combination of core-shell construction with dendritic morphology to enhance the catalytic performance for a given chemical reaction [118]. They firstly synthesized Au nanoparticles with well-defined shapes, e.g., cube, rod, or octahedron in aqueous solution, and then conducted the ascorbic acid reduction of K2PtCl4 in the presence of preformed Au seeds for the formation of heterogeneous Au-Pt bimetallic nanostructures with an Au core and a branched Pt shell. In specific, the final bimetallic Au-Pt nanostructures maintain the original morphologies of Au seeds, and exhibit enhanced but shape-dependent activities for ORR, suggesting the important role of the cores in boosting the core-shell particles for catalytic reactions.
Starting from Ag seeds with multi-twinned defects on their particle surfaces, Liu et al. developed an overgrowth strategy to produce heterogeneous core-shell Ag@Pt nanoparticles with an Ag core and a stellated Pt shell, which can combine the electronic effect between the Ag core and Pt shell due to their different electronegativities with the exposure of more active edges and corners due to the stellated morphologies to engineer the catalytic behaviors of Pt [119]. As schematically shown in Fig. 8, the core-shell Ag–Pt nanoparticles with an Ag core and a stellated Pt shells can be conveniently synthesized in oleylamine using an one-pot approach. In this strategy, multi-twinned Ag seeds are formed in advance due to fast reduction kinetics of Ag+ precursors in oleylamine, and then anisotropic growth of Pt on the Ag seeds occurs due to faster growth rate of Pt at the high-energy twin boundary sites than that at other places without defects. In the presence of sufficient Pt2+ precursors, extended Pt branches are formed on the twinned Ag seeds, which constitute the stellated shells in the final heterogeneous core-shell Ag–Pt products.

|
Download:
|
| Fig. 8. Schematic illustration to show the formation mechanism of core-shell Ag–Pt nanoparticles with stellated Pt shells in a one-pot synthesis. Copied with permission [119]. Copyright 2014, the Springer-Nature. | |
{kind=link}
Figs. 9a and b show the TEM and HRTEM images of the core-shell Ag–Pt nanoparticles as-prepared by reducing Ag+ and Pt2+ precursors in an one-pot approach, in which well-dispersed nanoparticles with stellated morphologies and an overall average size of 25.2 nm are clearly observed. In particular, the twinned feature of Ag seeds, which is not stable in nature, allows the internal structure of the stellated core-shell particles to be further modified [119]. By aging the stellated core-shell Ag–Pt nanoparticles with aqueous BSPP solution for 48 h at ambient conditions, the Ag cores in the stellated core-shell Ag–Pt nanoparticles can be completely eliminated, leaving behind the stellated Pt nanoparticles with hollow interiors, as shown by Figs. 9c and d for their TEM and HRTEM images, which definitely show that the removal of Ag cores does not induce the collapse of the stellated morphologies.

|
Download:
|
| Fig. 9. TEM images (a, c) and HRTEM images (b, d) of the heterogeneous core-shell Ag–Pt nanoparticles with stellated morphologies (a, b) and stellated Pt nanoparticles with hollow interiors (c, d), respectively. Copied with permission [119]. Copyright 2014, the Springer-Nature. | |
{kind=link}
It was noteworthy that core-shell Ag–Pt nanoparticles with stellated morphologies are highly active in catalyzing MOR due to the electronic coupling between Ag core and Pt shells, while stellated Pt nanoparticles with hollow interiors are active for ORR due to the abundant atomic steps, edges and corner atoms in their extended branches [119]. Therefore, the heterogeneous stellated particles provide an effective platform for tailoring their catalytic feasibility by changing the internal structures.
7. Heterogeneous dimers composed of Ag and hollow structured Pt nanoparticlesHeterogeneous dimers composed of two different metals might have unique physical and chemical properties due to the electronic interaction or lattice strain effect between their two constituent metals. For example, Zhang et al. modified the Pt nanoparticles with Au clusters through electrodeposition to form Au-Pd heterogeneous dimers, and they found the Au modification can significantly enhance the stability of Pt nanoparticles in electrochemically catalyzing ORR at room temperature [120]. When Au clusters connect with the Pt nanoparticles, they would modify the electronic configuration of the same and lower their surface energy or d-band state, favorable for preventing Pt nanoparticles from dissolution under potential cycling regimes.
Although some methods based on controlled overgrowth [107, 110], high-temperature reduction [121-123], post-synthesis modification [124], plasmon-mediated synthesis [112], kinetics regulation [125], seed-mediated reduction [126] and selective transformation [127], have been developed for producing nanosized bimetallic dimmers, the reports on the synthesis of heterogeneous dimers composed of two different metals are still very scarce. In addition, the structural control of each domain in the bimetallic dimers is also a grand challenge, which may greatly affect the physical or chemical properties of the heterogeneously bimetallic dimmers.
Using core-shell Ag–Pt nanoparticles as precursors, Liu et al. demonstrated a facile strategy with a high-yield to synthesize heterogeneously bimetallic dimers composed of Ag nanoparticles (Ag NPs) and hollow structured Pt nanoparticles, labeled as Ag-hPt dimeric nanoparticles (Ag-hPt dNPs) [128]. This strategy involves the first synthesis of core-shell Ag–Pt nanoparticles in oleylamine as precursors and the subsequent conversion of them into bimetallic Ag-hPt dNPs through the diffusion of Ag in core-shell Ag–Pt nanoparticles from inside to outside. A heating treatment at 80 ℃ was used to promote the inside-out diffusion of Ag in the core-shell precursors. As schematically illustrated by Fig. 10, at a suitable temperature, the Ag+ ions generated by O2-etching diffuse out from core-shell Ag–Pt nanoparticles and are reduced again by oleylamine to form single crystalline Ag nanoparticles on the outer surface of the Pt shell. The Ag nanoparticles grow with more diffused Ag+ ions until the Ag core completed disappears. Finally, the Ag nanoparticles on the surface of the Pt shell undergoes a ripening process to form larger and more stable single domain on the surface of Pt shell, leading to the formation of bimetallic Ag-hPt dNPs.

|
Download:
|
| Fig. 10. Schematic illustration showing the synthesis of heterogeneously bimetallic Ag-hollow Pt dimers via the diffusion of Ag in core-shell Ag–Pt nanoparticles from inside to outside. Copied with permission [128]. Copyright 2014, the Royal Society of Chemistry. | |
{kind=link}
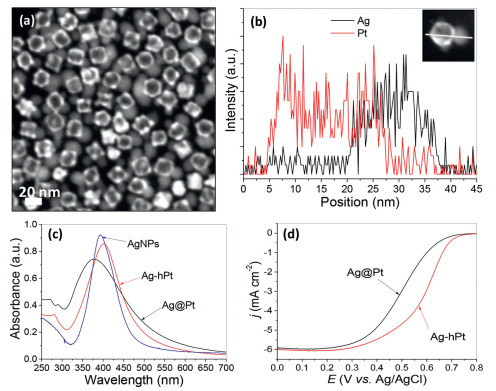
Fig. 11a shows the scanning TEM (STEM) image of the as-prepared Ag-hPt dNPs, in which the strong imaging contrast in Ag and hollow Pt domains are clearly observed. The formation of Ag-hPt dNPs can be directly proved by the line scanning analysis of a single dimeric particle under high-angle annular dark-field STEM mode (Fig. 11b), in which the distribution of Ag and Pt signals at two different sides are presented.

|
Download:
|
| Fig. 11. STEM image (a) and elemental profiles (b) of bimetallic Ag-hPt dNPs synthesized via the inside-out diffusion of Ag in core-shell Ag–Pt nanoparticles. UV–visible spectra recorded from organic suspensions of the core-shell Ag–Pt nanoparticles, bimetallic Ag-hPt dNPs and the sole Ag seeds (c). ORR polarization curves for core-shell Ag–Pt nanoparticles and bimetallic Ag-hPt dNPs, recorded at room temperature in an O2-saturated HClO4 solution (0.1 mol/L) at a sweep rate of 20 mV/s and a rotating speed of 1600 rpm (d). Copied with permission [128]. Copyright 2014, the Royal Society of Chemistry. | |
{kind=link}
As expected, the as-prepared bimetallic Ag-hPt dNPs exhibit much different optical and catalytic properties from those of their core-shell Ag–Pt counterparts. As indicated by Fig. 11c, uniformly coating a Pt shell on the Ag seeds would result in blue-shift of the surface plasmon resonance (SPR) of Ag, while asymmetric incorporation of Pt with Ag seeds (Ag-hPt dNPs) leads to red-shift of their SPR, suggesting that the optical property of Ag nanoparticles could be tuned by forming different nanostructures with a second metal. On the other hand, Fig. 11d shows that the Ag-hPt dNPs are much more active than their core-shell Ag–Pt counterparts for ORR, suggesting that relegating Ag to the core or to the shell region has a non-negligible impact on the Pt catalytic property. The conversion from core-shell Ag–Pt to bimetallic Ag-hPt heterogeneous dimers not only offers a method to design the structure of one metal domain in heterogeneous dimers, but also provides a typical example to illustrate tuning of the material properties by means of a structural tailoring.
8. Heterogeneous nanocomposites derived from core-shell Ag–Pt nanoparticlesMotivated by the huge potentials in electrocatalysis after the first report on using nanocomposites consisting of Ag2S and Pt to promote the electrooxidation of methanol [129], the synthesis of Pt-containing semiconductor-noble metal nanocomposites have received much attention in recent years [24, 25, 43-45]. These Pt-based heterogeneous nanocomposites are attractive in electrocatalytic applications. In brief, the adjacent semiconductor domains, which have different electron affinity from Pt metal, could change the electron density around the Pt atoms by interacting with the Pt domain through the interface in the nanocomposites. The changes in Pt electron density alter the adsorption/desorption of reactants/products on the Pt atoms, thus tuning their catalytic property in electrochemical reactions. In this sense, the catalytic performance of Pt domain for an electrochemical reaction might be dramatically enhanced by increasing the complexity of the heterogeneous Pt-based nanocomposites.
Starting with core-shell Ag–Pt nanoparticles as initial templates, Liu et al. reported a cost-effective strategy to prepare heterogeneous nanocomposites composed of Ag2S and hollow structured Pt nanoparticles [130]. Their original purpose is to find a cheap chemical agent, e.g., sulfur (S) that could replace expensive BSPP for preparing noble metal nanoparticles with hollow interiors. However, they experimentally found that when using sulfur to promote the diffusion of Ag in core-shell Ag–Pt nanoparticles from inside to outside, dimeric nanocomposites consisting of Ag2S and hollow Pt domains are dominantly formed instead of pure hollow structured Pt nanoparticles, as shown by Figs. 12a and b for their TEM and HRTEM images, in which the Ag2S domain and hollow structured Pt domain could be easily discerned by the brightness contrasts.

|
Download:
|
| Fig. 12. TEM (a) and HRTEM images (b) of Ag2S-hollow Pt nanocomposites as-prepared by reacting core-shell Ag–Pt nanoparticles with element sulfur. Reproduced with permission [119]. Copyright 2014, the Royal Society of Chemistry. TEM (c) and HRTEM images (d) of Ag2Se-hollow Pt nanocomposites as-prepared by reacting core-shell Ag–Pt nanoparticles with active Se species. Copied with permission [132]. Copyright 2016, the Elsevier. | |
{kind=link}
The synthesis of Ag2S-hollow Pt nanocomposites from core-shell Ag–Pt nanoparticles can be extended to prepare nanocomposites composed of other chalcogenide semiconductors and hollow structured Pt nanoparticles. For example, by replacing element sulfur with highly active hydrophobic Se species that formed in oleylamine by reacting Se powder with sodium borohydride (NaBH4) [131], Cui et al. synthesized nanocomposites composed of Ag2Se and hollow Pt nanoparticles from core-shell Ag–Pt precursors [132], which have analogous dimeric structure as that of Ag2S-hollow Pt nanocomposites, as evinced by Figs. 12c and d for their TEM and HRTEM images.
The protocol used to synthesize Ag2S-hollow Pt nanocomposites can be further extended to fabricate ternary heterogeneous nanocomposites composed of Ag2S, Au, and hollow structured Pt nanoparticles, labeled as Ag2S-Au-hPt [133]. Briefly, the as-prepared Ag2S-hollow Pt nanocomposites were mixed in toluene with Au3+ ions that transferred from aqueous solution. Then after aging the mixture for 2 h, Ternary Ag2S-Au-hPt nanocomposites are formed as the dominant product, as confirmed by the TEM and HRTEM images in Fig. 13, in which isolated Au nanoparticles are not found. Also, Au deposits only on the surface of Ag2S domains, indicating that under experimental conditions, the semiconductor section in the Ag2S-hollow Pt heterogeneous dimers are preferential sites for the nucleation and growth of the Au domain.

|
Download:
|
| Fig. 13. TEM (a) and HRTEM images (b) of ternary Ag2S-Au-hPt nanocomposites as-prepared by reacting Ag2S-hollow Pt dimers with Au3+ ions in toluene at room temperature. (c) Energy level diagram for ternary Ag2S-Au-hPt nanocomposites showing the intraparticle charge transfer among different domains. (d) Cyclic voltammograms of ternary Ag2S-Au-hPt nanocomposites and their core-shell Ag–Pt as well as Ag2S-hollow Pt dimers in 0.1 mol/L HClO4 with 1 mol/L methanol at a scan rate of 20 mV/s. Copied with permission [133]. Copyright 2014, the Springer-Nature. | |
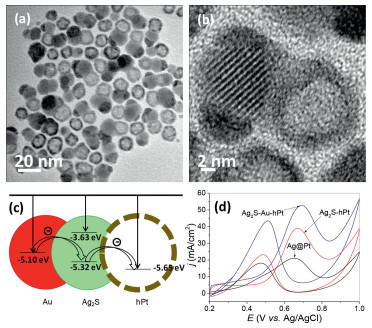{kind=link}
The strong electronic coupling among Ag2S, Au and Pt domains due to the energy level alignment (Fig. 13c) inevitably induce the increase in electron density of the Pt domains, which endow them with much higher electrocatalytic property for MOR than those of their core-shell Ag–Pt and dimeric Ag2S-hollow Pt counterparts, as evinced by Fig. 13d. The study offers a vivid example to demonstrate that the property of a material can be enhanced by a hybrid strategy, and the concept is definitely useful for designing and synthesizing heterogeneous nanostructures for more catalytic applications.
9. Conclusion and outlooksIn summary, we introduced a number of heterogeneous nanostructures synthesized using core-shell Ag–Pt nanoparticles as starting materials, including hollow, dimeric and composite structures and also highlighted their application in catalyzing methanol oxidation and oxygen reduction as electrocatalysts. Based on structural design or electronic interaction, these heterogeneous nanostructures show enhanced catalytic properties either in methanol oxidation or in oxygen reduction. This review also shows the capability of core-shell Ag–Pt nanoparticles in producing various heterogeneous nanostructures as starting templates. However, as we presented in this review, the average diameter of the core-shell Ag–Pt starting templates is ca. 14 nm, and this results in a big overall size for the derived heterogeneous nanostructures, not optimum for electrocatalytic applications. Rationally, using Ag nanoclusters as seeds to prepare core-shell Ag–Pt starting templates might be a feasible route to derive heterogeneous nanostructures with smaller sizes and higher catalytic performances. In addition, the heterogeneous nanostructures derived from core-shell Ag–Pt nanoparticles may serve as templates for preparing heterogeneous nanomaterials with more structural complexity that would boost their performance in a given catalytic reaction. The schemes in Fig. 14 show a number of possible routes for forming more complicated heterogeneous nanostructures using core-shell Ag–Pt nanoparticles or their derivatives as starting templates: (i) Starting with core-shell Ag–Pt nanoparticles, conducting galvanic replacement reaction with Au3+ ions in the core region of core-shell Ag–Pt nanoparticles so as to obtain heterogeneous Pt shells with an alloy Ag/Au shell inside; (ii) also starting with core-shell Ag–Pt nanoparticles, the difference from (i) is Pd2+ are used to replace Au3+ so that heterogeneous Pt shells with an alloy Ag/Pd shell inside could be obtained; (iii) starting with heterogeneous Ag-hollow Pt dimers, then conducting galvanic replacement reaction with Au3+ ions to obtain heterogeneous dimers composed of hollow Pt and hollow Ag/Au domains; (iv) also starting with heterogeneous Ag-hollow Pt dimers, replacing Au3+ ions with Pd2+ ions so as to obtain heterogeneous dimers composed of hollow Pt and hollow Ag/Pd domains. Of course, there must be other possible ways capable to generate heterogeneous nanostructures with optimal sizes and various domains that endow the heterogeneous nanostructures with desired characteristics for fulfilling application requirements.

|
Download:
|
| Fig. 14. Schematic illustrations to show the synthesis of heterogeneous nanostructures using core-shell Ag–Pt or their derivatives as staring templates. | |
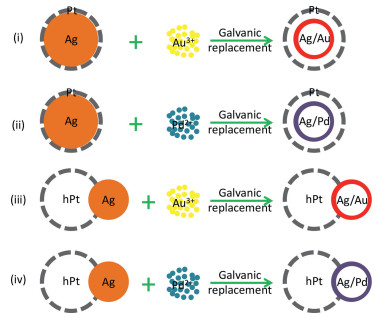{kind=link}
The authors declare no conflict of interest.
AcknowledgmentsFinancial supports from the Beijing Natural Science Foundation (No. Z200012), National Natural Science Foundation of China (Nos. 22075290, 21972068, 21776292 and 21706265), State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences (No. MPCS-2019-A-09), and Nanjing IPE Institute of Green Manufacturing Industry are gratefully acknowledged.
| [1] |
C.B. Murray, C.R. Kagan, M.G. Bawendi, Annu. Rev. Mater. Sci. 30 (2000) 545-610. DOI:10.1146/annurev.matsci.30.1.545 |
| [2] |
C. Burda, X. Chen, R. Narayanan, M.A. El-Sayed, Chem. Rev. 105 (2005) 1025-1102. DOI:10.1021/cr030063a |
| [3] |
S. Laurent, D. Forge, M. Port, et al., Chem. Rev. 108 (2008) 2064-2110. DOI:10.1021/cr068445e |
| [4] |
J. Chen, B. Lim, E.P. Lee, Y. Xia, Nano Today 4 (2009) 81-95. DOI:10.1016/j.nantod.2008.09.002 |
| [5] |
Y. Xia, Y. Xiong, B. Lim, S.E. Skrabalak, Angew. Chem. Int. Ed. 48 (2009) 60-103. DOI:10.1002/anie.200802248 |
| [6] |
N.T.K. Thanh, N. Maclean, S. Mahiddine, Chem. Rev. 114 (2014) 7610-7630. DOI:10.1021/cr400544s |
| [7] |
R. Jin, C. Zeng, M. Zhou, Y. Chen, Chem. Rev. 116 (2016) 10346-10413. DOI:10.1021/acs.chemrev.5b00703 |
| [8] |
L. Liu, A. Corma, Chem. Rev. 118 (2018) 4981-5079. DOI:10.1021/acs.chemrev.7b00776 |
| [9] |
A. Heuer-Jungemann, N. Feliu, I. Bakaimi, et al., Chem. Rev. 119 (2019) 4819-4880. DOI:10.1021/acs.chemrev.8b00733 |
| [10] |
I. Favier, D. Pla, M. Gómez, Chem. Rev. 120 (2020) 1146-1183. DOI:10.1021/acs.chemrev.9b00204 |
| [11] |
P.D. Cozzoli, T. Pellegrino, L. Manna, Chem. Soc. Rev. 35 (2006) 1195-1208. DOI:10.1039/b517790c |
| [12] |
L. Carbone, P.D. Cozzoli, Nano Today 5 (2010) 449-493. DOI:10.1016/j.nantod.2010.08.006 |
| [13] |
J. Yang, J.Y. Lee, J.Y. Ying, Chem. Soc. Rev. 40 (2011) 1672-1696. DOI:10.1039/B916790K |
| [14] |
R.G. Chaudhuri, S. Paria, Chem. Rev. 112 (2012) 2373-2433. DOI:10.1021/cr100449n |
| [15] |
N. Gao, X. Feng, Chem. Rev. 115 (2015) 8294-8343. DOI:10.1021/cr400607y |
| [16] |
M. Ha, J.H. Kim, M. You, et al., Chem. Rev. 119 (2019) 12208-12278. DOI:10.1021/acs.chemrev.9b00234 |
| [17] |
M. Sankar, Q. He, R.V. Engel, et al., Chem. Rev. 120 (2020) 3890-3938. DOI:10.1021/acs.chemrev.9b00662 |
| [18] |
C. Yang, F. Lv, Y. Zhang, et al., Adv. Energy Mater. 9 (2019) 1902674. DOI:10.1002/aenm.201902674 |
| [19] |
Z. Chen, Y. Liu, C. Liu, et al., Small 16 (2020) 1904964. DOI:10.1002/smll.201904964 |
| [20] |
L. Zong, X. Chen, S. Dou, et al., Chin. Chem. Lett. (2020). DOI:10.1016/j.cclet.2020.08.029 |
| [21] |
C. Liu, Z. Chen, D. Rao, et al., Sci. China Mater. 64 (2021) 611-620. |
| [22] |
J. Shi, Chem. Rev. 113 (2013) 2139-2181. DOI:10.1021/cr3002752 |
| [23] |
A. Sitt, I. Hadar, U. Banin, Nano Today 8 (2013) 494-513. DOI:10.1016/j.nantod.2013.08.002 |
| [24] |
J. Qu, F. Ye, D. Chen, et al., Adv. Colloid Interface Sci. 230 (2016) 29-53. DOI:10.1016/j.cis.2015.12.010 |
| [25] |
J. Yang, Wiley-VCH, Weinheim, 2019.
|
| [26] |
X. Liu, C. Lee, W.C. Law, et al., Nano Lett. 13 (2013) 4333-4339. DOI:10.1021/nl402124h |
| [27] |
V. Mazumder, M. Chi, K.L. More, S. Sun, J. Am. Chem. Soc. 132 (2010) 7848-7849. DOI:10.1021/ja1024436 |
| [28] |
H. Ataee-Esfahani, L. Wang, Y. Nemoto, Y. Yamauchi, Chem. Mater. 22 (2010) 6310-6318. DOI:10.1021/cm102074w |
| [29] |
M. Peng, J. Hu, H. Zeng, J. Am. Chem. Soc. 132 (2010) 10771-10785. DOI:10.1021/ja102105q |
| [30] |
J.H. Yang, J. Yang, J.Y. Ying, ACS Nano 6 (2012) 9373-9382. DOI:10.1021/nn303298s |
| [31] |
Q. Zhang, I. Lee, J.B. Joo, F. Zaera, Y. Yin, Acc. Chem. Res. 46 (2013) 1816-1824. DOI:10.1021/ar300230s |
| [32] |
X. Xia, Y. Wang, A. Ruditskiy, Y. Xia, Adv. Mater. 25 (2013) 6313-6333. DOI:10.1002/adma.201302820 |
| [33] |
W. van der Stam, E. Bladt, F.T. Rabouw, S. Bals, C. De Mello Donega, ACS Nano 9 (2015) 11430-11438. DOI:10.1021/acsnano.5b05496 |
| [34] |
D. Zhang, Y. Yang, Y. Bekenstein, et al., J. Am. Chem. Soc. 138 (2016) 7236-7239. DOI:10.1021/jacs.6b03134 |
| [35] |
B.A. Koscher, N.D. Bronstein, J.H. Olshansky, Y. Bekenstein, A.P. Alivisatos, J. Am. Chem. Soc. 138 (2016) 12065-12068. DOI:10.1021/jacs.6b08178 |
| [36] |
A.G.M. Da Silva, T.S. Rodrigues, S.J. Haigh, P.H.C. Camargo, Chem. Commun. 53 (2017) 7135-7148. DOI:10.1039/C7CC02352A |
| [37] |
M. Lin, G.H. Kim, J.H. Kim, J.W. Oh, J.M. Nam, J. Am. Chem. Soc. 139 (2017) 10180-10183. DOI:10.1021/jacs.7b04202 |
| [38] |
J. Ahn, D. Wang, Y. Ding, J. Zhang, D. Qin, ACS Nano 12 (2018) 298-307. DOI:10.1021/acsnano.7b06353 |
| [39] |
C. Bianchini, P.K. Shen, Chem. Rev. 109 (2009) 4183-4206. DOI:10.1021/cr9000995 |
| [40] |
A. Chen, P. Holt-Hindle, Chem. Rev. 110 (2010) 3767-3804. DOI:10.1021/cr9003902 |
| [41] |
U. Banin, Y. Ben-Shahar, K. Vinokurov, Chem. Mater. 26 (2013) 97-110. |
| [42] |
D. Chen, P. Cui, H. Liu, J. Yang, Sci. Adv. Today 1 (2015) 25225. |
| [43] |
H. Liu, Y. Feng, D. Chen, et al., J. Mater. Chem. A 3 (2015) 3182-3223. DOI:10.1039/C4TA05801A |
| [44] |
J. Tang, D. Chen, Q. Yao, J. Xie, J. Yang, Mater. Today Energy 6 (2017) 115-127. DOI:10.1016/j.mtener.2017.09.005 |
| [45] |
D. Liu, L. Xu, J. Xie, J. Yang, Nano Mater. Sci. 1 (2019) 184-197. DOI:10.1016/j.nanoms.2019.03.005 |
| [46] |
V. Bansal, A.P. O'Mullane, S.K. Bhargava, Electrochem. Commun. 11 (2009) 1639-1642. DOI:10.1016/j.elecom.2009.06.018 |
| [47] |
F. Ye, H. Liu, W. Hu, et al., Dalton Trans. 41 (2012) 2898-2903. DOI:10.1039/c2dt11960a |
| [48] |
W. Zhang, J. Yang, X. Lu, ACS Nano 6 (2012) 7397-7405. DOI:10.1021/nn302590k |
| [49] |
M. Tsuji, M. Hamasaki, A. Yajima, et al., Mater. Lett. 121 (2014) 113-117. DOI:10.1016/j.matlet.2014.01.093 |
| [50] |
P. Duchesne, P. Zhang, J. Phys. Chem. C 118 (2014) 21714-21721. DOI:10.1021/jp507382z |
| [51] |
D. Ma, X. Tang, M. Guo, H. Lu, X. Xu, Ionics 21 (2015) 1417-1426. DOI:10.1007/s11581-014-1290-1 |
| [52] |
S. Schaefer, F. Muench, E. Mankel, et al., Nano 10 (2015) 1550085. DOI:10.1142/S179329201550085X |
| [53] |
D. Chen, F. Ye, H. Liu, J. Yang, Sci. Rep. 6 (2016) 24600. DOI:10.1038/srep24600 |
| [54] |
T. Fu, J. Fang, C. Wang, J. Zhao, J. Mater. Chem. A 4 (2016) 8803-8811. DOI:10.1039/C6TA02202B |
| [55] |
T. Fu, J.-.X. Huang, S. Lai, et al., J. Power Sour. 365 (2017) 17-25. DOI:10.1016/j.jpowsour.2017.08.066 |
| [56] |
Y. Zhang, J. Li, H. Rong, X. Tong, Z. Wang, Langmuir 33 (2017) 5991-5997. DOI:10.1021/acs.langmuir.7b00647 |
| [57] |
S. Kumar-Krishnan, M. Estevez-González, R. Pérez, R. Esparza, M. Meyyappan, RSC Adv. 7 (2017) 27170-27176. DOI:10.1039/C7RA04301E |
| [58] |
X. Sun, X. Yang, Y. Zhang, et al., Nanoscale 9 (2017) 15107-15114. DOI:10.1039/C7NR04366J |
| [59] |
Y. Ma, X. Wu, G. Zhang, Appl. Catal. B: Environ. 205 (2017) 262-270. DOI:10.1016/j.apcatb.2016.12.025 |
| [60] |
T. Wang, J. Zhou, Y. Wang, Nanomaterials 8 (2018) 331. DOI:10.3390/nano8050331 |
| [61] |
S.R. Dash, S.S. Bag, A.K. Golder, Sens. Actuat. B: Chem. 314 (2020) 128062. DOI:10.1016/j.snb.2020.128062 |
| [62] |
U. Aslam, S. Linic, ACS Appl. Mater. Interfaces 9 (2017) 43127-43132. DOI:10.1021/acsami.7b14474 |
| [63] |
H. Liu, J. Qu, Y. Chen, et al., J. Am. Chem. Soc. 134 (2012) 11602-11610. DOI:10.1021/ja302518n |
| [64] |
P. Hou, P. Cui, H. Liu, J. Li, J. Yang, Nano Res. 8 (2015) 512-522. DOI:10.1007/s12274-014-0663-0 |
| [65] |
D. Xu, P. Xu, X. Wang, et al., ACS Appl. Mater. Interfaces 12 (2020) 8091-8097. DOI:10.1021/acsami.9b17201 |
| [66] |
P. Hou, H. Liu, J. Li, J. Yang, CrystEngComm 17 (2015) 1826-1832. DOI:10.1039/C4CE02560A |
| [67] |
C. Wang, F. Ye, C. Liu, H. Cao, J. Yang, Colloids Surf. A 385 (2011) 85-90. DOI:10.1016/j.colsurfa.2011.05.051 |
| [68] |
J. Yang, J.Y. Ying, J. Am. Chem. Soc. 132 (2010) 2114-2115. DOI:10.1021/ja909078p |
| [69] |
B. Wiley, T. Herricks, Y. Sun, Y. Xia, Nano Lett. 4 (2004) 1733-1739. DOI:10.1021/nl048912c |
| [70] |
H. Liang, H. Zhang, J. Hu, et al., Angew. Chem. Int. Ed. 43 (2004) 1540-1543. DOI:10.1002/anie.200352956 |
| [71] |
J. Yang, J.Y. Lee, H.-.P. Too, S. Valiyaveettil, J. Phys. Chem. B 110 (2006) 125-129. DOI:10.1021/jp055306c |
| [72] |
F. Caruso, R.A. Caruso, H. Möhwald, Science 282 (1998) 1111-1114. DOI:10.1126/science.282.5391.1111 |
| [73] |
F. Caruso, X. Shi, R.A. Caruso, A. Susha, Adv. Mater. 13 (2001) 740-744. DOI:10.1002/1521-4095(200105)13:10<740::AID-ADMA740>3.0.CO;2-6 |
| [74] |
Z. Yang, Z. Niu, Y. Lu, Z. Hu, C.C. Han, Agnew. Chem. Int. Ed. 115 (2003) 1987-1989. DOI:10.1002/ange.200250443 |
| [75] |
X.W. Lou, C. Yuan, L.A. Archer, Small 3 (2007) 261-265. DOI:10.1002/smll.200600445 |
| [76] |
Z. Wang, D. Luan, F.Y.C. Boey, X.W. Lou, J. Am. Chem. Soc. 133 (2011) 4738-4741. DOI:10.1021/ja2004329 |
| [77] |
X. Lai, J. Li, B.A. Korgel, et al., Angew. Chem. Int. Ed. 50 (2011) 2738-2741. DOI:10.1002/anie.201004900 |
| [78] |
F. Bai, Z. Sun, H. Wu, et al., Nano Lett. 11 (2011) 3759-3762. DOI:10.1021/nl201799x |
| [79] |
H.T. Schmidt, A.E. Ostafin, Adv. Mater. 14 (2002) 532-535. DOI:10.1002/1521-4095(20020404)14:7<532::AID-ADMA532>3.0.CO;2-4 |
| [80] |
L. Qi, J. Li, J. Ma, Adv. Mater. 14 (2002) 300-303. DOI:10.1002/1521-4095(20020219)14:4<300::AID-ADMA300>3.0.CO;2-P |
| [81] |
D. Zhang, L. Qi, J. Ma, H. Cheng, Adv. Mater. 14 (2002) 1499-1502. DOI:10.1002/1521-4095(20021016)14:20<1499::AID-ADMA1499>3.0.CO;2-5 |
| [82] |
M.S. Wong, J.N. Cha, K.-.S. Choi, T.J. Deming, G. D Stucky, Nano Lett. 2 (2002) 583-587. DOI:10.1021/nl020244c |
| [83] |
D.H.W. Hubert, M. Jung, A.L. German, Adv. Mater. 12 (2000) 1291-1294. DOI:10.1002/1521-4095(200009)12:17<1291::AID-ADMA1291>3.0.CO;2-Z |
| [84] |
X. Gao, J. Zhang, L. Zhang, Adv. Mater. 14 (2002) 290-293. DOI:10.1002/1521-4095(20020219)14:4<290::AID-ADMA290>3.0.CO;2-U |
| [85] |
T. Nakashima, N. Kimizuka, J. Am. Chem. Soc. 125 (2003) 6386-6387. DOI:10.1021/ja034954b |
| [86] |
H.M. Chen, R.-.S. Liu, M.-.Y. Lo, et al., J. Phys. Chem. C 112 (2008) 7522-7526. |
| [87] |
Z. Peng, J. Wu, H. Yang, Chem. Mater. 22 (2009) 1098-1106. |
| [88] |
Y.-.N. Tan, J. Yang, J.Y. Lee, D.I.C. Wang, J. Phys. Chem. C 111 (2007) 14084-14090. DOI:10.1021/jp0741049 |
| [89] |
H. Liu, F. Ye, J. Yang, Ind. Eng. Chem. Res. 53 (2014) 5925-5931. DOI:10.1021/ie5002718 |
| [90] |
W.-.M. Zhang, J.-.S. Hu, Y.-.G. Guo, et al., Adv. Mater. 20 (2008) 1160-1165. DOI:10.1002/adma.200701364 |
| [91] |
X.W. Lou, C.M. Li, L.A. Archer, Adv. Mater. 21 (2009) 2536-2539. DOI:10.1002/adma.200803439 |
| [92] |
J.S. Chen, C.M. Li, W.W. Zhou, et al., Nanoscale 1 (2009) 280-285. DOI:10.1039/b9nr00102f |
| [93] |
J.S. Chen, D. Luan, C. M, et al., Chem. Commun. 46 (2010) 8252-8254. DOI:10.1039/c0cc02973d |
| [94] |
J.S. Chen, Z. Wang, X.C. Dong, P. Chen, X.W. Lou, Nanoscale 3 (2011) 2158-2161. DOI:10.1039/c1nr10162e |
| [95] |
J. Lee, J.C. Park, H. Song, Adv. Mater. 20 (2008) 1523-1528. DOI:10.1002/adma.200702338 |
| [96] |
D.V. Gough, A. Wolosiuk, P.V. Braun, Nano Lett. 9 (2009) 1994-1998. DOI:10.1021/nl900264n |
| [97] |
J. Liu, S.Z. Qiao, S.B. Hartono, G.Q. Lu, Angew. Chem. Int. Ed. 49 (2010) 4981-4985. DOI:10.1002/anie.201001252 |
| [98] |
J. Gao, G. Liang, B. Zhang, et al., J. Am. Chem. Soc. 129 (2007) 1428-1433. DOI:10.1021/ja067785e |
| [99] |
W. Zhao, H. Chen, Y. Li, et al., Adv. Funct. Mater. 18 (2008) 2780-2788. DOI:10.1002/adfm.200701317 |
| [100] |
Y. Zhu, T. Ikoma, N. Hanagata, S. Kaskel, Small 6 (2010) 471-478. DOI:10.1002/smll.200901403 |
| [101] |
J. Zhong, C. Cao, H. Liu, Y. Ding, J. Yang, Ind. Eng. Chem. Res. 52 (2013) 1303-1308. DOI:10.1021/ie302652b |
| [102] |
F. Benabid, F. Couny, J.C. Knight, T.A. Birks, P.J. Russell, Nature 434 (2005) 488-491. DOI:10.1038/nature03349 |
| [103] |
K. Kamata, Y. Lu, Y. Xia, J. Am. Chem. Soc. 125 (2003) 2384-2385. DOI:10.1021/ja0292849 |
| [104] |
H. Li, Z. Bian, J. Zhu, et al., J. Am. Chem. Soc. 129 (2007) 8406-8407. DOI:10.1021/ja072191c |
| [105] |
Y. Feng, F. Ye, H. Liu, J. Yang, Sci. Rep. 5 (2015) 16219. DOI:10.1038/srep16219 |
| [106] |
S. Zhou, K. Mcllwrath, G. Jackson, B. Eichhorn, J. Am. Chem. Soc. 128 (2006) 1780-1781. DOI:10.1021/ja056924+ |
| [107] |
H. Lee, S.E. Habas, G.A. Somorjai, P. Yang, J. Am. Chem. Soc. 130 (2008) 5406-5407. DOI:10.1021/ja800656y |
| [108] |
Z. Peng, H. Yang, Nano Today 4 (2009) 143-164. DOI:10.1016/j.nantod.2008.10.010 |
| [109] |
K.D. Beard, D. Borrelli, A.M. Cramer, et al., ACS Nano 3 (2009) 2841-2853. DOI:10.1021/nn900214g |
| [110] |
B. Lim, H. Kobayashi, T. Yu, et al., J. Am. Chem. Soc. 132 (2010) 2506-2507. DOI:10.1021/ja909787h |
| [111] |
F. Wang, C. Li, L.-.D. Sun, et al., J. Am. Chem. Soc. 133 (2011) 1106-1111. DOI:10.1021/ja1095733 |
| [112] |
M.R. Langille, J. Zhang, C.A. Mirkin, Angew. Chem. Int. Ed. 50 (2011) 3543-3547. DOI:10.1002/anie.201007755 |
| [113] |
L.F. Zhang, S.L. Zhong, A.W. Xu, Angew. Chem. Int. Ed. 52 (2013) 645-649. DOI:10.1002/anie.201205279 |
| [114] |
B. Lim, M. Jiang, P.H.C. Camargo, et al., Science 324 (2009) 1302-1305. DOI:10.1126/science.1170377 |
| [115] |
Z. Peng, H. Yang, J. Am. Chem. Soc. 131 (2009) 7542-7543. DOI:10.1021/ja902256a |
| [116] |
S. Guo, S. Dong, E. Wang, ACS Nano 4 (2010) 547-555. DOI:10.1021/nn9014483 |
| [117] |
L. Wang, Y. Nemoto, Y. Yamauchi, J. Am. Chem. Soc. 133 (2011) 9674-9677. DOI:10.1021/ja202655j |
| [118] |
Y. Kim, J.W. Hong, Y.W. Lee, et al., Angew. Chem. Int. Ed. 49 (2010) 10197-10201. DOI:10.1002/anie.201005839 |
| [119] |
H. Liu, F. Ye, Q. Yao, et al., Sci. Rep. 4 (2014) 3969. |
| [120] |
J. Zhang, K. Sasaki, E. Sutter, R.R. Adzic, Science 315 (2007) 220-222. DOI:10.1126/science.1134569 |
| [121] |
T. Pellegrino, A. Fiore, E. Carlino, et al., J. Am. Chem. Soc. 128 (2006) 6690-6698. DOI:10.1021/ja0607741 |
| [122] |
J.-.S. Choi, H.J. Choi, D.C. Jung, J.-.H. Lee, J. Cheon, Chem. Commun. (2008) 2197-2199. |
| [123] |
C. Wang, W. Tian, Y. Ding, et al., J. Am. Chem. Soc. 132 (2010) 6524-6529. DOI:10.1021/ja101305x |
| [124] |
Z. Peng, H. Yang, Nano Res. 2 (2009) 406-415. DOI:10.1007/s12274-009-9040-9 |
| [125] |
J. Zeng, C. Zhu, J. Tao, et al., Angew. Chem. Int. Ed. 51 (2012) 2354-2358. DOI:10.1002/anie.201107061 |
| [126] |
S.I. Lim, M. Varon, I. Ojea-Jiménez, J. Arbiol, V. Puntes, J. Mater. Chem. 21 (2011) 11518-11523. DOI:10.1039/c1jm10313j |
| [127] |
L. Bai, Y. Kuang, J. Luo, D.G. Evans, X. Sun, Chem. Commun. 48 (2012) 6963-6965. DOI:10.1039/c2cc32557h |
| [128] |
H. Liu, J. Yang, J. Mater. Chem. A 2 (2014) 7075-7081. DOI:10.1039/C4TA00243A |
| [129] |
J. Yang, J.Y. Ying, Angew. Chem. Int. Ed. 50 (2011) 4637-4643. DOI:10.1002/anie.201101213 |
| [130] |
H. Liu, F. Ye, H. Cao, et al., Nanoscale 5 (2013) 6901-6907. DOI:10.1039/c3nr01949g |
| [131] |
Y. Wei, J. Yang, A.W.H. Lin, J.Y. Ying, Chem. Mater. 22 (2010) 5672-5677. DOI:10.1021/cm101308f |
| [132] |
P. Cui, H. He, H. Liu, S. Zhang, J. Yang, J. Power Sour. 327 (2016) 432-437. DOI:10.1016/j.jpowsour.2016.07.079 |
| [133] |
Y. Feng, H. Liu, P. Wang, et al., Sci. Rep. 4 (2014) 6204. |