 2021, Vol. 32
2021, Vol. 32 


b Sinopec Beijing Research Institute of Chemical Industry, Beijing 100013, China
With the increasing serious energy and environmental issues, water splitting has been considered as a promising technology to make H2 energy [1]. Water splitting involves two half-reactions: H2 evolution reaction (HER) and O2 evolution reaction (OER). Compared to the HER, the effectiveness of OER is hindered due to sluggish kinetics. At present, precious metal oxides (i.e., IrO2, and RuO2) are effective electrocatalysts for OER [2]. However, the low reserve and high price dramatically limit their widespread application [3]. Therefore, researchers focus on the development of transition-metal-based materials [4-10]. Cobalt (Co)-based materials were regarded as efficient water oxidation catalysts owing to their rich content, low price, and excellent performance[11-13]. Novel methodologies have been proposed to develop highly efficient Co-based catalysts [14-16]. Noted that hierarchical structures have advantages of high permeability, high conductivity and rich active sites, which are benefit for electrocatalytic OER [17-19]. However, electrocatalytic activities of these Co-based hierarchical materials are normally limited by their compositions.
Co-based catalysts used for OER mainly includes: Co-N-C materials [20-22], Co nitrides [23], Co oxides/hydroxides [24-26], Co perovskites [27], Co chalcogenides [28], Co selenides [29], Co phosphides [30, 31], Co phosphates[32], and Co phosphosulfides [33], etc. For example, Hua et al. reported a hybrid CoP/Co2P material, which exhibited efficient overall water splitting activity with a cell voltage of 1.57 V at j = 10 mA/cm2 [34]. Wang et al. found that the introduction of S2- into the Co-based hydroxide could tune the electronic and surface structure and lead to enhanced OER performance [35]. It is usually believed that Co atom is the real active site for OER and the anions such as S and P could modify the electronic structure of metal Co [36, 37]. Though great contributions have been dedicated to developing advanced OER materials, revealing the relationship between anion species and electrocatalytic activities still remains a great challenge. Furthermore, comprehensive investigation into electronic structures and catalytic properties of hierarchical Co-based materials with the same morphology and size is still lacking.
Herein, we synthesized Co-A (A = O, Se, P) hexagrams from the same hierarchical Co(OH)2/Co(OH)F precursor. The oxidation/selenation/phosphorization processes do not change the morphology and size of the precursor, which is important to exclude their influence. In addition, hierarchical structures not only can help the diffusion process but also expose more active sites. Electrochemical measurements show that Co-P hexagram exhibits well OER performance with an overpotential of 269 mV, outperforming Co-O (399 mV) and Co-Se (347 mV) at j = 10 mA/cm2 in 1.0 mol/L KOH. By combining the theoretical calculation, the P substitution could modulate the electronic structure of Co.
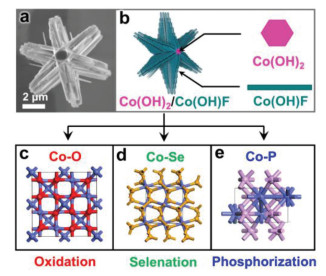
The Co precursor was first synthesized using hydrothermal synthesis. The powder X-ray diffraction (PXRD) pattern demonstrates that the obtained material is a mixture of Co(OH)2 (JCPDS No. 30-0443) and Co(OH)F (JCPDS No. 50-0827) (Fig. S1 in Supporting information). Scanning electron microscope (SEM) image shows that Co precursor has a novel hexagram-like morphology with six branches surrounded on a hexagonal core (Fig. 1). In particular, each branch consists of uniform nanorods with a width of ~200 nm and a length ranging from 1 μm to 3 μm (Fig. 1a). During the formation process, hexagonal Co(OH)2 core first forms, and then Co(OH)F nanorod grows along hexagon edges due to matched unit cell parameters. The specific growth mechanism has been studied in details in our previous work [38]. Therefore, Co precursor is constructed with a two-dimensional (2D) Co(OH)2 as the internal core and one-dimensional (1D) Co(OH)F nanorods as external branches (Fig. 1b).

|
Download:
|
| Fig. 1. SEM image (a) and structural illustration (b) of Co precursor, and (c-e) crystal structures of Co-A (A = O, Se, P) hexagrams. | |
{kind=link}
Co-A (A = O, Se, P) hexagrams were prepared by the oxidation/selenation/phosphorization process at the same temperature (350 ℃). Crystal structures of Co-A (A = O, Se, P) hexagrams are shown in Figs. 1c–e. Characteristic peaks in PXRD patterns of Co-A (A = O, Se, P) hexagrams indicate that these materials are Co3O4 (JCPDS No. 43-1003), CoSe2 (JCPDS No. 09-0234) and CoP (JCPDS No. 29-0497), respectively (Fig. S2 in Supporting information). Co-O hexagrams were obtained by heating precursors at the muffle furnace under air. Herein, the structure of Co-O hexagrams (Co3O4) is different from previous reported CoO hexagrams, which were obtained by heating the precursor under Ar [15]. Co-Se and Co-P hexagrams were prepared by heating Se powder and NaH2PO2·H2O at the upstream of pipe furnace under Ar flow, respectively. The unique and novel hexagram-like morphology of Co precursor remains after the oxidation, selenation or phosphorization process, which is important to further discuss their electrochemical properties (Fig. S3 in Supporting information).
Transmission electron microscope (TEM) images of Co precursor and Co-A (A = O, Se, P) hexagrams are displayed in Fig. 2. TEM and high-resolution TEM (HRTEM) images of Co precursor demonstrate that the crystal lattice has a space of 0.26 nm, which is attributed to the (011) face of Co(OH)F (Figs. 2a–c). The Co3O4 branch shows perfect crystal lattice with a spacing of 0.29 nm corresponding to the (200) face of Co3O4 (Figs. 2d–f). The crystal lattice with a d-spacing of 0.24 nm, which is assigned to the (211) facet of CoSe2, confirming the formation of the CoSe2 phase (Figs. 2g–i). The HRTEM image of Co-P shows an interplanar distance of 0.25 nm, which is indexed to the (111) plane of CoP (Figs. 2j–l). Co-O hexagrams exhibit a Brunauer-Emmett-Teller surface area of 17.3 m2/g and an average pore size of ~ 25 nm (Fig. S4 in Supporting information). These catalysts have similar surface area due to the same morphology.

|
Download:
|
| Fig. 2. TEM and HRTEM images of Co(OH)2/Co(OH)F (a-c), Co3O4 (d–f), CoSe2 (g–i), and CoP (j–l) hexagrams. | |
{kind=link}
X-ray photoelectron spectroscopy (XPS) spectra measurements of Co-A (A = O, Se, P) hexagrams were performed to confirm valence states of surface elements (Fig. S5 in Supporting information). Peaks appeared at 779.7 and 781.3 eV can be indexed to the main photoelectron lines from Co3+ and Co2+ ions in Co3O4, respectively (Fig. 3a) [39, 40]. Peaks appeared at 784.7 and 789.7 eV are attributed to their satellite shake-up peaks [41, 42]. In the XPS spectrum of O 1s of Co3O4, three peaks were observed (Fig. S5b). The peak appeared at 529.9 eV is attributed to the lattice oxygen, while the peak appeared at 531.8 eV is ascribed to oxygen vacancies [16]. Another peak appeared at 533.4 eV corresponds to hydroxyl species of surface-adsorbed water molecules. For Co-Se hexagram, the peak of Co 2p3/2 appeared at 778.4 eV is corresponded to Co2+ cations in Co-Se hexagrams (Fig. S5c). Peaks of Se 3d at 54.2 and 55.2 eV are also in good agreement with CoSe2 (Fig. S5d) [43]. Co 2p XPS spectrum of Co-P hexagrams shows a peak appeared at 778.5 eV indexing to the Co 2p3/2 of Co species in Co-P hexagrams (Fig. S5e). Peaks appeared at 782.4 and 786.7 eV are well indexed to the Co 2p3/2 of oxidized Co species. The peaks for P 2p at 129.8 and 131.4 eV are well matched with the P3- (Fig. S5f). The predominate peak appeared at 135.2 eV is assigned to PO43- [44]. XPS results indicate that Co-A (A = O, Se, P) hexagrams with the similar morphology and size possess different chemical environments, which may further affect their electrocatalysis.

|
Download:
|
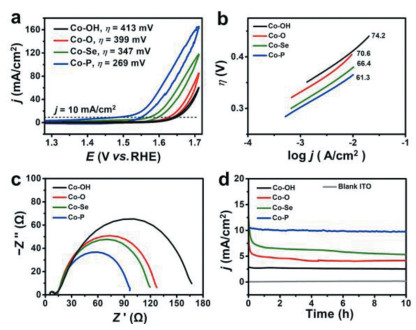
| Fig. 3. CV data (a), Tafel plots (b), EIS (c) and controlled potential electrolysis (d) of Co-A (A = O, Se, P) hexagrams. | |
{kind=link}
Electrochemical cyclic voltammetry (CV) data of Co(OH)2/Co(OH)F (Co-OH) and Co-A (A = O, Se, P) hexagrams were measured in 1.0 mol/L KOH solution at 0.05 V/s (Fig. 3a). Co-P hexagrams show the smallest overpotential (η) of 269 mV compared to Co-OH (η = 413 mV), Co-O (η = 399 mV) and Co-Se hexagrams (η = 347 mV) at j = 10 mA/cm2, demonstrating the excellent OER performance of Co-P hexagrams. Co-P hexagrams exhibit good OER performance compared to other reported Co-based catalysts (Table S1 in Supporting information). In addition, Co-P hexagrams show an obvious CoⅡ/CoⅢ couple at ~1.12 V (vs. reversible hydrogen electrode, RHE) compared to Co-Se and Co-O hexagrams (Fig. S6 in Supporting information). This is because Co atoms in Co-P hexagrams are easily oxidized due to relatively larger number of valence electron as confirmed with the theoretical calculation. Considering the full oxidation of CoⅢ/CoⅣ event at ~1.46 V (vs. RHE) and CoⅣ/CoⅢ event at ~1.44 V (vs. RHE) in the cathodic return scan for Co-O hexagrams, a -CoⅤ=O formal oxidation state might be formed, which involves the cleavage/formation of O-O [17]. In contrast, Co-P and Co-Se hexagrams start to oxidize water with the occurring of CoⅢ/CoⅣ event (Fig. S6). Therefore, Co species in Co-P and Co-Se hexagrams are more active than that in Co-O hexagrams. Linear sweep voltammetry (LSV) data further confirm this result (Fig. S7 in Supporting information). Herein, the OER performance of Co-O hexagrams (Co3O4, η = 399 mV) is much lower than that of previous reported CoO hexagrams (CoO, η = 269 mV). This is because CoO hexagrams have abundant oxygen defects and long-term ordering when pyrolyzed at critical temperature (400 ℃) under Ar [15]. Tafel plots of Co-A (A = O, Se, P) hexagrams were utilized to study their OER kinetics (Fig. 3b). Co-P hexagrams have the smallest Tafel slope (61.3 mV/dec) compared to Co-OH precursor (74.2 mV/dec), Co-O hexagrams (70.6 mV/dec) and Co-Se hexagrams (66.4 mV/dec), indicating the rapid OER rates. These similar Tafel values demonstrated that Co-A hexagrams had the same rate-limiting step for OER process. The improved catalytic activity of Co-P may be attributed to the enhanced charge and mass transfer. Electrochemical impedance spectroscopy (EIS) results indicate that Co-P hexagrams have the smallest charge-transfer resistance compared to Co-O and Co-Se hexagrams (Fig. 3c). Therefore, the P substitution increases the electrical conductivity compared to O and Se anions.
To further understand the improved water oxidation activity, we measured the electrochemical double-layer capacitance (Cdl) to calculate the electrochemical surface area (Fig. S8 in Supporting information) [45]. The Cdl of Co-P hexagrams (6.8 mF/cm2) is much larger than that of Co-Se (2.4 mF/cm2) and Co-O hexagrams (1.0 mF/cm2), respectively (Fig. S9 in Supporting information). Therefore, Co-P hexagrams expose more active sites. The controlled potential electrolysis (CPE) of Co-A (A = O, Se, P) hexagrams is tested at 1.63 V (vs. RHE) with the indium tin oxide (ITO) electrode. Co-P hexagrams show well stability at j = ~10 mA/cm2 during the CPE of 10 h compared to Co-Se and Co-O hexagrams (Fig. 3d).
Redox peaks of CV current-potential responses for Co-P hexagrams appeared at ~1.05 and ~1.50 V (vs. RHE) are attributed to the oxidation of CoⅡ/CoⅢ and CoⅢ/CoⅣ, respectively (Fig. S7)[17]. Linear i-ν1/2 responses of Co-A (A = O, Se, P) hexagrams indicate their diffusion-controlled kinetics in OER (Figs. S10–S12 in Supporting information). The linear correlation of potentials (E1 and E2) vs. pH proves the process of proton-coupled electron transfer (Figs. S10d–f). For Co-Se and Co-O hexagrams, similar linear correlations were also observed (Figs. S11 and S12 in Supporting information).
XPS results of Co 2p, O 1s, Se 3d, and P 2p for Co-A (A = O, Se, P) hexagrams after CPE are shown in Fig. S13 (Supporting information). Main XPS peaks of Co 2p3/2 of Co3O4 are almost unchanged, while the appearance of a shoulder peak appeared at 530.2 eV of O 1 s indicates that there are surface Co oxide species, i.e., CoOOH, after CPE (Figs. S13a and b) [17]. The main peaks of Co 2p of CoSe2 still remain after CPE (Fig. S13c), while a broad peak of SeOx at 60.4 eV is observed after electrolysis (Fig. S10d) [43]. For Co-P hexagram, an additional peak at 780.5 eV ascribed to Co oxide appears (Fig. S13e) [46, 47]. Oxidized species were observed in Co-A (A = O, Se, P) hexagrams, indicating that they all experienced oxidation processes during the OER. HRTEM images after OER test further confirm these results (Fig. S14 in Supporting information). Therefore, the effects of O, Se and P on the intrinsic structures of Co and the OER activity cannot be ignored.
To further understand electronic structures, we calculated total density of states (TDOS) and projected DOS (PDOS) of Co-A (A = O, Se, P) are shown in Fig. 4.

|
Download:
|
| Fig. 4. TDOS and PDOS calculated for the Co-O (a), Co-Se (b) and Co-P (c). Black dashed horizontal line represents the Fermi level. | |
{kind=link}
The existence of intrinsic band gap and the mergence of Fermi levels into the conduction band indicate that both Co-O and Co-Se undergo a transition from semiconducting to metallic state. The Co-P displays a zero-band gap, i.e., a metallic nature, which is beneficial for the electron transfer process. Furthermore, a strong hybridization throughout a wide energy range is found between Co-d and O-p/Se-p states, showing a strong covalency of the bonding in Co-O/Co-Se; while P-p state in Co-P only has an insignificant influence on the overall DOS, which retains the metallic property of Co. Therefore, the Co-P has the rapid charge-transfer kinetic compared to Co-O/Co-Se, which agrees well with the experimental charge-transfer resistance (Fig. 3c).
In addition, a Bader analysis of the charge density was performed for understanding the interplay between Co cation and anions (O, Se, and P). The obtained valence electron number of Co is 7.92, 8.64, and 8.83 for Co-O, Co-Se, and Co-P, respectively. Thus, it can be inferred that the valence electron donated by Co in Co-P is less than that in Co-O or Co-Se. Thus, the interaction between Co and P is weaker than the interactions between Co and O, or between Co and Se, which is in accordance with the DOS analyses. In addition, It has been reported that the formation of adsorbed *OOH species is the rate-limiting step of OER [48]. Therefore, the interaction between adsorbed *OOH species and 3d orbital of transition metal determines the resulting OER activity. The theoretical calculation demonstrates that the increased 3d orbital electron density of Co-based materials favors the formation of adsorbed *OOH species, thus enhancing OER activity[49]. Herein, the metal Co in Co-P has the largest valence electron number (8.83) compared to Co-O (7.92) and Co-Se (8.64). Therefore, the Co-P is beneficial for the formation of adsorbed *OOH species and exhibits the best OER performance. Theoretical calculation demonstrated that the introduction of P could tune the electronic structure of Co compared to O and Se and then improve the charge transfer and lead to the easier generation of higher average oxidation state of Co species during OER process. Therefore, Co cation in Co-P hexagram is easier to be oxidized experimentally, when performing CV measurements (Fig. S6).
In conclusion, hierarchical Co-A hexagrams (A = O, Se, P) with the same morphology and size were prepared by oxidation, selenation, and phosphorization of Co precursors, respectively. Co-P hexagrams display the highest OER efficiency compared to Co-O and Co-Se hexagrams. The introduction of O, Se and P could effectively change the intrinsic electronic structure of Co and then accelerate the charge transfer. Furthermore, the P institution leads to the easier generation of active Co species during OER process, which might be ascribed to the metallic nature of Co-P hexagrams and the weak interaction between Co and P. This work offers a fundamental understanding of anions induced electrocatalytic properties of hierarchical Co-based materials.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for support from the National Natural Science Foundation of China (Nos. 21808138, 21773146, 21975148 and 21601118), Fok Ying-Tong Education Foundation for Outstanding Young Teachers in University, China Postdoctoral Science Foundation (No. 2019T120877), Fundamental Research Funds for the Central Universities (No. GK202103029), and Young Talent Fund of University Association for Science and Technology in Shaanxi, China (No. 20200602).
Appendix A. Supplementary dataSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.04.051.
| [1] |
W. Zhang, W. Lai, R. Cao, Chem. Rev. 117 (2017) 3717-3797. DOI:10.1021/acs.chemrev.6b00299 |
| [2] |
B. Liu, C. Wang, Y. Chen, Electrochim. Acta 264 (2018) 350-357. DOI:10.1016/j.electacta.2018.01.141 |
| [3] |
W. Xia, A. Mahmood, Z. Liang, R. Zou, S. Guo, Angew. Chem. Int. Ed. 55 (2016) 2650-2676. DOI:10.1002/anie.201504830 |
| [4] |
S. Anantharaj, S.R. Ede, K. Sakthikumar, et al., ACS Catal. 6 (2016) 8069-8097. DOI:10.1021/acscatal.6b02479 |
| [5] |
B. Wang, X. Cui, J. Huang, R. Cao, Q. Zhang, Chin. Chem. Lett. 29 (2018) 1757-1767. DOI:10.1016/j.cclet.2018.11.021 |
| [6] |
F. Cao, G. Pan, Y. Zhang, X. Xia, Chin. Chem. Lett. 31 (2020) 2230-2234. DOI:10.1016/j.cclet.2020.01.037 |
| [7] |
H. Wang, S. Zhu, J. Deng, et al., Chin. Chem. Lett. 32 (2020) 291-298. |
| [8] |
P. Han, T. Tan, F. Wu, et al., Chin. Chem. Lett. 31 (2020) 2469-2472. DOI:10.1016/j.cclet.2020.03.009 |
| [9] |
L. Zeng, L. Yang, J. Lu, et al., Chin. Chem. Lett. 29 (2018) 1875-1878. DOI:10.1016/j.cclet.2018.10.026 |
| [10] |
X. Zhao, J. Meng, Z. Yan, F. Cheng, J. Chen, Chin. Chem. Lett. 30 (2019) 319-323. DOI:10.1016/j.cclet.2018.03.035 |
| [11] |
J. Wang, W. Cui, Q. Liu, et al., Adv. Mater. 28 (2016) 215-230. DOI:10.1002/adma.201502696 |
| [12] |
H. Osgood, S.V. Devaguptapu, H. Xu, J. Cho, G. Wu, Nano Today 11 (2016) 601-625. DOI:10.1016/j.nantod.2016.09.001 |
| [13] |
Y. Wang, C. Yang, Y. Huang, et al., J. Mater. Chem. A 8 (2020) 6699-6708. DOI:10.1039/D0TA00010H |
| [14] |
Z. Xiao, Y. Wang, Y.C. Huang, et al., Energy Environ. Sci. 10 (2017) 2563-2569. DOI:10.1039/C7EE01917C |
| [15] |
Z. Liang, Z. Huang, H. Yuan, et al., Chem. Sci. 9 (2018) 6961-6968. DOI:10.1039/C8SC02294A |
| [16] |
T. Ling, D.Y. Yan, Y. Jiao, et al., Nat. Commun. 7 (2016) 12876. DOI:10.1038/ncomms12876 |
| [17] |
S. Wan, J. Qi, W. Zhang, et al., Adv. Mater. 29 (2017) 1700286. DOI:10.1002/adma.201700286 |
| [18] |
K. Yan, Z. Liang, Z. Li, et al., CrystEngComm 20 (2018) 5249-5255. DOI:10.1039/C8CE01234B |
| [19] |
G. Cai, W. Zhang, L. Jiao, S.H. Yu, H.L. Jiang, Chem 2 (2017) 791-802. DOI:10.1016/j.chempr.2017.04.016 |
| [20] |
Z. Liang, C. Zhang, H. Yuan, et al., Chem. Commun. 54 (2018) 7519-7522. DOI:10.1039/C8CC02646G |
| [21] |
Y. Jia, Y. Wang, G. Zhang, et al., J. Energy Chem. 49 (2020) 283-290. DOI:10.1016/j.jechem.2020.01.034 |
| [22] |
Z. Liang, X. Fan, H. Lei, et al., Angew. Chem. Int. Ed. 57 (2018) 13187-13191. DOI:10.1002/anie.201807854 |
| [23] |
Y. Zhang, B. Ouyang, J. Xu, et al., Angew. Chem. Int. Ed. 55 (2016) 8670-8674. DOI:10.1002/anie.201604372 |
| [24] |
Z. Liang, C. Zhang, Y. Xu, et al., ACS Sustain. Chem. Eng. 7 (2018) 3527-3535. |
| [25] |
Y. Tong, H. Liu, M. Dai, L. Xiao, X. Wu, Chin. Chem. Lett. 31 (2020) 2295-2299. DOI:10.1016/j.cclet.2020.03.029 |
| [26] |
Y. Wang, C. Yang, Z. Li, Z. Liang, G. Cao, Small 16 (2020) 2001973. DOI:10.1002/smll.202001973 |
| [27] |
S. Zhou, X. Miao, X. Zhao, et al., Nat. Commun. 7 (2016) 11510. DOI:10.1038/ncomms11510 |
| [28] |
Y. Yan, Y. Mengwei, L. Huifeng, S. Genban, M. Shulan, Electrochim. Acta 281 (2018) 198-207. DOI:10.1016/j.electacta.2018.05.182 |
| [29] |
X. Zhao, X. Li, Y. Yan, et al., Appl. Catal. B: Environ. 236 (2018) 569-575. DOI:10.1016/j.apcatb.2018.05.054 |
| [30] |
M. Song, Z. Zhang, Q. Li, et al., J. Mater. Chem. A 7 (2019) 3697-3703. DOI:10.1039/C8TA10985K |
| [31] |
J. Shi, F. Qiu, W. Yuan, M. Guo, Z.H. Lu, Chem. Eng. J. 403 (2021) 126312. DOI:10.1016/j.cej.2020.126312 |
| [32] |
L. Xie, R. Zhang, L. Cui, et al., Angew. Chem. Int. Ed. 56 (2017) 1064-1068. DOI:10.1002/anie.201610776 |
| [33] |
Z. Liang, Z. Yang, J. Dang, et al., Chem. Eur. J. 25 (2019) 621-626. |
| [34] |
Y. Hua, Q. Xu, Y. Hu, et al., J. Energy Chem. 37 (2019) 1-6. DOI:10.1016/j.jechem.2018.11.010 |
| [35] |
B. Wang, C. Tang, H.F. Wang, et al., Adv. Mater. 31 (2019) 1805658. DOI:10.1002/adma.201805658 |
| [36] |
P. Ge, C. Zhang, H. Hou, et al., Nano Energy 48 (2018) 617-629. DOI:10.1016/j.nanoen.2018.04.018 |
| [37] |
Z. Fang, L. Peng, Y. Qian, et al., J. Am. Chem. Soc. 140 (2018) 5241-5247. DOI:10.1021/jacs.8b01548 |
| [38] |
Z. Liang, Z. Yang, Z. Huang, et al., Electrochim. Acta 271 (2018) 526-536. DOI:10.1016/j.electacta.2018.03.186 |
| [39] |
Z. Chen, C.X. Kronawitter, B.E. Koel, Phys. Chem. Chem. Phys. 17 (2015) 29387-29393. DOI:10.1039/C5CP02876K |
| [40] |
L. Zhuang, L. Ge, Y. Yang, et al., Adv. Mater. 29 (2017) 1606793. DOI:10.1002/adma.201606793 |
| [41] |
J. Yang, H. Liu, W.N. Martens, R.L. Frost, J. Phys. Chem. C 114 (2010) 111-119. DOI:10.1021/jp908548f |
| [42] |
M.C. Biesinger, B.P. Payne, A.P. Grosvenor, et al., Appl. Surf. Sci. 257 (2011) 2717-2730. DOI:10.1016/j.apsusc.2010.10.051 |
| [43] |
D. Kong, H. Wang, Z. Lu, Y. Cui, J. Am. Chem. Soc. 136 (2014) 4897-4900. DOI:10.1021/ja501497n |
| [44] |
B. Qiu, L. Cai, Y. Wang, et al., Adv. Funct. Mater. 28 (2018) 1706008. DOI:10.1002/adfm.201706008 |
| [45] |
C.C.L. McCrory, S. Jung, J.C. Peters, T.F. Jaramillo, J. Am. Chem. Soc. 135 (2013) 16977-16987. DOI:10.1021/ja407115p |
| [46] |
J. Chang, Y. Xiao, M. Xiao, et al., ACS Catal. 5 (2015) 6874-6878. DOI:10.1021/acscatal.5b02076 |
| [47] |
N. Jiang, B. You, M. Sheng, Y. Sun, Angew. Chem. Int. Ed. 54 (2015) 6251-6254. DOI:10.1002/anie.201501616 |
| [48] |
L. Hui, Y. Xue, D. Jia, et al., Adv. Energy Mater. 8 (2018) 1800175. DOI:10.1002/aenm.201800175 |
| [49] |
T. Tang, W.J. Jiang, S. Niu, et al., J. Am. Chem. Soc. 139 (2017) 8320-8328. DOI:10.1021/jacs.7b03507 |