 2021, Vol. 32
2021, Vol. 32 


b State Environmental Protection Key Lab of Environmental Risk Assessment and Control on Chemical Processes, School of Resources & Environmental Engineering, East China University of Science and Technology, Shanghai 200237, China;
c Shanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China;
d College of Resources and Environmental Sciences, Nanjing Agricultural University, Nanjing 210095, China
Advanced oxidation process (AOPs) can be used as tertiary treatment strategies for the decontamination of organic micro-pollutants that are widely present in municipal wastewater treatment plants effluents [1]. Among different AOPs, sulfate radical (SO4·−) based AOPs (SR-AOPs) have attracted increasing scientific attention in recent years, due to their strong oxidizing ability, high selectivity and wide pH flexibility against numerous organic micro-pollutants [2, 3]. SO4·− could be generated by activated peroxymonosulfate (PMS, HSO5‒) or persulfate (PS, S2O82‒) via various approaches such as heating, UV irradiation, transition metals, bases and ultrasound [4-6]. Owing to the advantages mentioned above, SR-AOPs have been widespread applied in the remediation of soil, surface water and groundwater as a promising in situ chemical oxidation (ISCO) technology [4, 7].
As one of the most important components in natural waters or wastewaters, it was found recently that dissolved oxygen (DO), might be involved in SR-AOPs and led to unpredicted results on the degradation kinetic as well as pathways [8, 9]. For instance, Zhu et al. [8] found that the degradation of hexachloroethane by thermo-activated persulfate (TAP) was faster under anaerobic condition than aerobic condition, mainly due to the formation of S2O8·− in the absence of oxygen (Eq. 1).
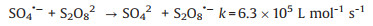
|
(1) |
The new formed S2O8− radical could continue react efficiently with halogenated organic compounds such as organochlorine pesticides by a reductive dechlorination process [8]. However, another theoretical study revealed a contradictory result that DO was beneficial to the degradation of pollutants, indeed Zhang et al. [10] showed that DO could decrease the energy barrier of the SR-AOPs and shifted the chemical equilibrium of the reaction towards the side of products. The conflicting literature results suggested the impact of DO on SR-AOPs might be diverse each other, depending on the target pollutants and reaction conditions. In addition, oxygen contents in different bodies are also variable, unlike surface environments, groundwater is often oxygen-deficient. Therefore, for realistic applications of SR-AOPs, it is of great importance to evaluate the impact of DO on the oxidation efficiency and degradation pathways of target pollutants since only few studies dealt with this point.
In our previous paper the degradation kinetic and transformation pathways of trimethoprim (TMP) were investigated systemically by thermo-activated persulfate (TAP) [11]. TMP is a bacteriostatic antibiotic for the treatment of infectious diseases in humans and have been frequently detected in natural waters [12, 13]. An oxygen involved pathway was temporarily proposed by which α-hydroxytrimethoprim (TMP−OH) and α-ketotrimethoprim (TMP = O) were generated simultaneously. However, the detailed impacts of oxygen on degradation kinetics and mechanisms still remain unclear.
Therefore, the main goal of the present study was to investigate the impact of oxygen on TMP oxidation more deeply. Our objectives were: 1) To evaluate the effect of oxygen on TMP degradation kinetic; 2) to identify the oxidation products and to record their evolution in the absence and presence of oxygen; 3) to reveal the detailed degradation pathways and mechanisms of TMP under different oxygen conditions.
Specifically, TAP oxidation experiments were conducted in 75 mL screw-cap cylindrical glass vials with Teflon septa at predetermined temperature (60 ℃) controlled by a thermal water bath (Xianou Instrument Manufacture Co., Ltd., Nanjing, China). Photochemical experiments were performed using a merry-go-round photo reactor (NaAi Instrument Manufacture Co., Ltd., Shanghai, China) equipped with a 500 W Xenon lamp and conducted in a series of 60 mL uncapped cylindrical quartz tubes. Appropriate volumes of TMP and PS stock solution were transferred into the vials to achieve a total of 50 mL reaction solution with predetermined concentrations. Anaerobic experiments were carried out in an anaerobic incubator (YQI-Ⅱ, CIMO, Shanghai, China), prior to activation, the reaction solutions were purged by N2 for 15 min to achieve a DO concentration of 1.28 ± 0.25 mg/L. Reactions solutions were buffered by using 10 mmol/L phosphate, pH values remained within 7.0 ± 0.1 over the course of the experiments. Sample aliquots (0.75 mL) were withdrawn intermittently and quenched by 20 μL Na2S2O3 (1 mol/L) solution to stop the reactions. Further analysis details are illustrated in Text S1 (Supporting information). Density functional theory (DFT) calculations were conducted in a Gaussian 09 package software [14]. Detailed calculation method was shown in Text S2 (Supporting information). In this contribution, frontier electron densities (FEDs) of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were also calculated. Values of 2FEDHOMO2 were estimated to predict reaction sites for electron-transfer by sulfate radicals [15].
Firstly, a comparison study was conducted to evaluate the impact of DO on TMP oxidation by sulfate radical. The theoretical DO concentration in an air-opened condition at 60 ℃ was around 3.18 mg/L and this system was taken as the control group. As seen in Fig. 1, in the case of control group, TMP underwent a fast degradation with a rate constant of 0.042 min−1. However, when oxygen was driven out from the reaction solution (anaerobic condition group), significant decrease on the degradation efficiency was observed, with a rate constant at 0.021 min−1. Correspondingly, half-life time of TMP was increased from 16.5 min to 33.0 min. TMP degradation under air-saturated condition was 2-fold faster than that at anaerobic condition.

|
Download:
|
| Fig. 1. Degradation of TMP under different oxygen conditions by TAP. ([TMP]0 = 40 μmol/L, [PS]0 = 2 mmol/L, T = 60 ℃, pH 7.0). | |
{kind=link}
To further confirm the involvement of oxygen, TMP degradation was also conducted in UV/PS process at ambient temperature (20 ℃), by continuously bubbling N2 (0.2 L/min) to achieve anaerobic conditions. Under control group (DO 8.23 ± 0.25 mg/L), TMP loss rate was detected at 0.033 min−1, which was faster than that of anaerobic condition group (DO 1.52 ± 0.20 mg/L) at 0.018 min-1 (Fig. S1 in Supporting information). All the results suggested that TMP degradation under control group was faster than that at anaerobic condition, indicating the involvement of DO in the overall interaction between sulfate radical and TMP.
It was widely considered sulfate radical and hydroxyl radical was responsible for the elimination of contaminants in the TAP processes [16, 17]. The work of Tan et al. has reported O2·− might be generated under the basic conditions in the antipyrine removal by TAP [18]. Thus, spin trapping-electron paramagnetic resonance (EPR) analysis was employed here to identify the potential involved active species. As shown in (Fig. S2 in Supporting information), under both two oxygen conditions, the splitting signals with relative intensity ratio of 1:2:2:1 and a six-line signal were observed, whose typical spectra were assigned to DMPO-·OH and DMPO-SO4·−, respectively [19, 20]. However, O2·− was hardly detected directly by EPR, possibly due to its low concentration. These phenomena confirmed TMP removal was mainly affected by ·OH and SO4·−.
To further explore the dominant species on TMP removal by TAP processes under different oxygen conditions, the radical scavenger experiments were conducted. The different reactivities of tertiary butanol (TBA) and ethanol (EtOH) toward hydroxyl radical ( k·OH+EtOH = 2.0 × 109 L mol−1 s-1, k·OH+TBA = 5.7 × 108 L mol−1 s-1) and sulfate radical ( kSO4·-+ EtOH = 4.65 × 107 L mol−1 s-1, kSO4·-+ TBA = 6.55 × 105 L mol−1 s-1) could be used to distinguish the dominant radical species in reaction solution [21, 22]. In the meanwhile, chloroform (CF) was applied to trap O2·− with rate constant of 3 × 1010 L mol−1 s-1 [21]. Applied in Fig. S3 (Supporting information), without the addition of any scavenger, 85.7% TMP removal could be achieved in the control group after 60 min. After the addition of EtOH, TMP removal sharply decreased from 85.7% to 26.3%. When TBA replaced EtOH, the decline of TMP was comparatively slighter. In addition, the presence of CF showed less adverse effect on TMP removal, indicating O2·− playing a minor role in the TMP oxidation. In the N2 bubbling group, same trend was also observed similarly. All these results suggested that overweight SO4·− was the predominant oxidizing species in the TAP process.
In order to obtain a deeper insight on the involved mechanism of DO in the reaction process, oxidation products generated under two different oxygen contents were identified by HR-MS technique. Surprisingly, no difference on the total ion chromatogram (TIC) spectrum of TMP degradation under different conditions was observed (Fig. S4 in Supporting information). Specifically, two oxidation products with m/z of 307.1396 and 305.1242 (Table S1 and Fig. S5 in Supporting information) were identified as the primary products, assigning as TMP−OH (λmax at 270.6 nm) and TMP = O (λmax at 226.8, 255.0 and 293.2 nm) (Fig. S6 in Supporting information), respectively, which was consistent with our previous work [11]. It has also been documented by Zhang et al. [23] that these two chemicals were the only transformation products in UV-activated PS induced oxidation of TMP in synthetic human urine. Therefore, the following discussion would be mainly based on this conclusion.
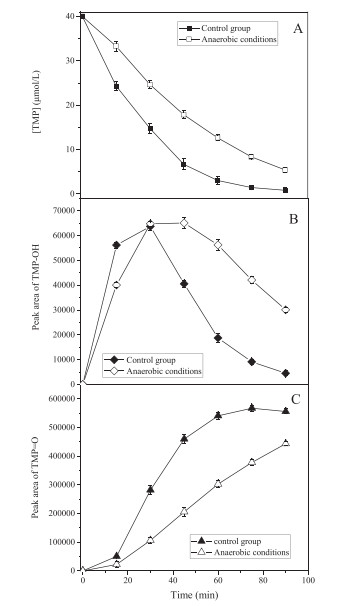
The mass results seemed contradictory to kinetic study in Fig. 1, since oxidation products were nearly the same in the absence and presence of DO. However, from the quantitative point of view, some differences were observed. The evolution of TMP−OH and TMP = O at different conditions were analyzed by HPLC-DAD (Fig. S6). As seen in Fig. 2, TMP−OH and TMP = O exhibited different formation profiles under control group and anaerobic conditions. Specifically, with the continuous degradation of TMP, TMP−OH reached its maximum concentrations at about 30 min in the presence of DO, afterwards, it begun to decay. A similar pattern was observed under anoxic condition with the maximum TMP−OH level occurred at 45 min. In the case of TMP = O, the evolution patterns were quite different, for control group sample, TMP = O exhibited a similar profile to TMP−OH and reached maximum around 75 min; while in anaerobic condition, TMP = O showed a continuous increasing tendency. It is noteworthy that over the experimental course the concentration of TMP = O in the absence of oxygen lower than that at the control group open to air. In addition, after the same conversion rate, the yields of TMP−OH and TMP = O in the absence of oxygen were higher than that under control group. For instance, after 15 min of reaction under control group, 40% of TMP was removed, which corresponds to the area of TMP−OH and TMP = O were 56163 (270 nm) and 50502 (293 nm), respectively (Fig. S6A). While in anaerobic condition, 30 min of reaction is required to obtain the same rate of 40% of conversion and the peak area of 64782 and 106653 for TMP−OH and TMP = O, respectively. This could be explained by the fact that in the presence of DO, TMP−OH and TMP = O might undergo further oxidation, leading to relative lower concentrations. In general, the discrepancies on oxidation products evolution profiles and distributions reflect the intrinsic differences of TMP reaction with sulfate radicals under different oxygen conditions.

|
Download:
|
| Fig. 2. Degradation of TMP (A) under control group (■) and anaerobic conditions (⬜). Time-dependent evolution of absorption peaks of TMP−OH (B) and TMP = O (C) in HPLC-DAD, TMP−OH (270 nm) under control group (◆) and anaerobic conditions (◇), TMP = O (293 nm) under control group (▲) and anaerobic conditions (△). ([TMP]0 = 40 μmol/L, [PS]0 = 2 mmol/L, T = 60 ℃, pH 7.0). | |
{kind=link}
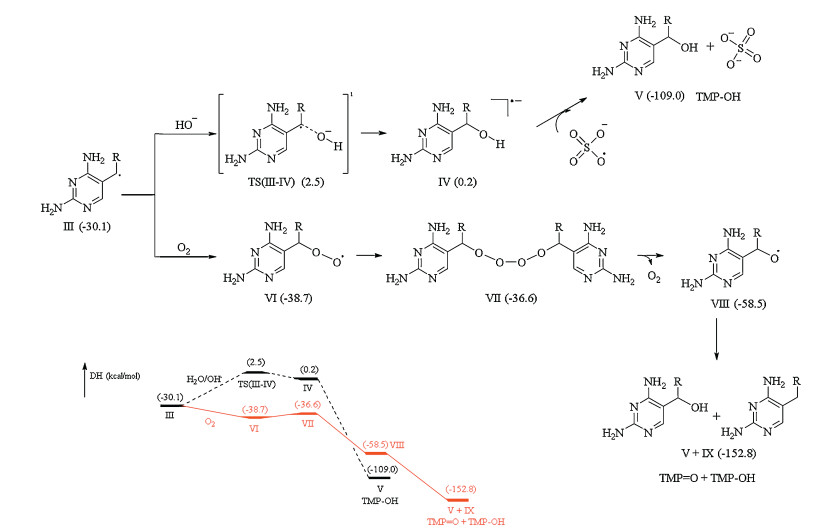
To better understand the underlying interaction mechanisms through which SO4·− reacts with TMP, the reactive sites were predicated by calculating FEDs values (Tables S2 and S3 in Supporting information). The result suggests that 5C (Scheme 1) of TMP opposes the highest 2FEDHOMO2 value, indicating that SO4·− preferentially attacks this position. Based on kinetic study, products identification, calculation results, as well as literature data, we attempted to propose the detailed oxidation pathways of TMP by TAP in the presence and absence of oxygen, as shown in Scheme 1. The initial step was believed to be an electron-transfer reaction from TMP to SO4·−, and a pyrimidine radical cation could be formed, which had been experimentally proved by EPR [24]. The pyrimidine radical cation is unstable and would undergo a deprotonation process from the bridging methylene carbon (−CH2−) to generate a carbon-centered radical. Afterwards, two different pathways were proposed in which one is oxygen dependent while the second one is oxygen independent. The first path was initialed via a quick combination with DO to form the corresponding peroxyl radical, followed by a peroxyl radical bimolecular combination to generate a tetroxide intermediate. Further unimolecular decomposition of tetroxide gives TMP−OH and TMP = O, which has been well accepted by the literature [25]. On the other way, the carbon-centered radical could also undergo hydrolysis to produce an OH-adduct radical, which can be further oxidized by another SO4·− to form TMP−OH, this process was considered as an oxygen independent pathway. Furthermore, TMP−OH could be further oxidized by SO4·−, undergoing a similar path to that from TMP to TMP−OH (without oxygen), as 5C was also the reactive site of TMP−OH (Table S3).

|
Download:
|
| Scheme 1. Proposed oxidation pathways for TMP degradation by TAP, the red line represents the oxygen dependent pathways. | |
{kind=link}
To further support our proposed oxidation pathways, DFT calculations were performed to elucidate the decisive involvement of DO in TMP oxidation processes, the calculation results are illustrated in Fig. 3, Fig. 4. Initially, we calculated the primary interaction between TMP and SO4·−. By considering the one electron-transfer process, the first step was the addition of SO4·− on C5 via a transition state (TS(Ⅰ-Ⅱ)) in Fig. 3), resulting in the generation of an adduct intermediate Ⅱ. For such process an energy barrier of 0.2 kcal/mol was obtained, which could be easily to overcome in TAP. The intermediate Ⅱ was unstable and could undergo spontaneous cleavage to give the carbon centered radical (Ⅲ) and HSO4‒ (or H+ and SO42‒), this process was expected to be thermodynamically favorable due to the relatively low energy level of Ⅲ. The proposed addition and dissociation mechanisms of sulfate radical have already been documented in literatures [26, 27]. Once the carbon centered radical was formed, there were two different pathways for the further transformation. When oxygen was present in the system, DO could combine with species Ⅲ to form corresponding peroxyl radical (species Ⅵ), this process was thermodynamically spontaneous due to the lower energy level of species Ⅵ. For the transformation from Ⅵ to Ⅶ, a small energy barrier of 2.1 kcal/mol was calculated, which was easy to overcome in our study. The following reaction (Ⅶ → Ⅷ) was also thermodynamically favorable, leading to the formation of TMP = O and TMP−OH, spontaneously. However, in the absence of oxygen system, the transformation of species Ⅲ would overcome a higher energy barrier compared to the system with oxygen. The hydroxylation could directly take place on 9C, leading to the formation of TMP−OH. Therefore, from a thermodynamical point of view, the oxygen dependent path was more favorable that the oxygen independent one, this could explain the promotion effect of DO on TMP decay in TAP.

|
Download:
|
| Fig. 3. The calculated enthalpy profiles (kcal/mol) for the reaction of TMP with sulfate radical. | |
{kind=link}

|
Download:
|
| Fig. 4. The calculated enthalpy profiles (kcal/mol) for the transformation pathways of carbon centered radical with sulfate radical in the presence and absence of DO. | |
{kind=link}
Based on the proposed mechanisms, the evolution profiles of TMP and its corresponding products (Fig. 2) could be further explained. With the oxidation of TMP, under control group, the formation of both TMP−OH and TMP = O at the initial stage (within 15 min) was faster than that at anaerobic condition. Such result was in line with the DFT calculation that TMP−OH/TMP = O could be formed via two different paths in the presence of oxygen. After 30 min, in the presence of DO, TMP−OH level begun to decay with a relative fast decomposition rate (30−60 min), which could be attributed to the fact that most TMP in the reaction system was consumed at this stage, the reaction between sulfate radical and TMP−OH would become crucial, leading to a fast formation of TMP = O. In the absence of DO, the reaction would follow the sequence of TMP → TMP−OH → TMP = O, leading to the continuous generation of TMP = O.
Total organic carbon (TOC) changes were also tracked under different oxygen environments. Applied in Fig. S7 (Supporting information), the values of TOC dropped drastically after 60 min under control group. However, in the anaerobic condition group, TOC remained quite stable. The presence of oxygen will promote the reduction of TOC, which was of great environmental significance for the application of SR-AOPs.
To conclude, from the experimental data and calculation results, DO was found to play an important role on the oxidation kinetic and pathways of TMP reaction with SO4·−. The absence of DO would decline the degradation rate constants, as well as alter the degradation pathways, although same oxidation products were detected. Without DO, TMP reacted with SO4·− through a hydroxylation process to produce TMP−OH, further oxidation of TMP−OH would generate TMP = O. When DO was involved, besides hydroxylation-oxidation process, a second pathway driven by DO would take place, leading to final formation TMP−OH and TMP = O, simultaneously.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgementsThis work was financially supported by the National Natural Science Foundation of China (No. 21806037). The authors would also like to thank Dr. Ting Wu from Research Center of Analysis and Test of East China University of Science and Technology for the technical support on HR-MS analysis.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.062.
| [1] |
R. Andreozzi, V. Caprio, A. Insola, R. Marotta, Catal. Today 53 (1999) 51-59. DOI:10.1016/S0920-5861(99)00102-9 |
| [2] |
R. Yuan, S.N. Ramjaun, Z. Wang, J. Liu, J. Hazard. Mater. 196 (2011) 173-179. DOI:10.1016/j.jhazmat.2011.09.007 |
| [3] |
W.D. Oh, Z. Dong, T.T. Lim, Appl. Catal B: Environ 194 (2016) 169-201. DOI:10.1016/j.apcatb.2016.04.003 |
| [4] |
A. Tsitonaki, B. Petri, M. Crimi, H. Mosbæk, R.L, et al., Environ. Sci. Tech. 40 (2010) 55-91. DOI:10.1080/10643380802039303 |
| [5] |
O.S. Furman, A.L. Teel, R.J. Watts, Environ. Sci. Tech. 44 (2010) 6423-6428. DOI:10.1021/es1013714 |
| [6] |
L. Zhou, C. Ferronato, J.M. Chovelon, et al., Chem. Eng. J. 311 (2017) 28-36. DOI:10.1016/j.cej.2016.11.066 |
| [7] |
G.P. Anipsitakis, D.D. Dionysiou, Environ. Sci. Technol. 37 (2003) 4790-4797. DOI:10.1021/es0263792 |
| [8] |
C. Zhu, F. Zhu, C. Liu, et al., Environ. Sci. Tech. 52 (2018) 8548-8557. DOI:10.1021/acs.est.7b06279 |
| [9] |
W. Qin, G. Fang, Y. Wang, D. Zhou, Chem. Eng. J. 348 (2018) 526-534. DOI:10.1016/j.cej.2018.04.215 |
| [10] |
R. Zhang, X. Wang, L. Zhou, et al., Water Res. 135 (2018) 144-154. DOI:10.1016/j.watres.2018.02.028 |
| [11] |
Y. Ji, W. Xie, Y. Fan, et al., Chem. Eng. J. 286 (2016) 16-24. DOI:10.1016/j.cej.2015.10.050 |
| [12] |
A.L. Batt, D.S. Aga, Anal. Chem. 77 (2005) 2940-2947. DOI:10.1021/ac048512+ |
| [13] |
A. Göbel, C.S. McArdell, M.J.F. Suter, et al., Anal. Chem. 76 (2004) 4756-4764. DOI:10.1021/ac0496603 |
| [14] |
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120 (2008) 215-241. DOI:10.1007/s00214-007-0310-x |
| [15] |
L. Zhou, Y. Zhang, R. Ying, et al., Environ. Sci. Pollut. Res. 24 (2017) 11549-11558. DOI:10.1007/s11356-017-8672-7 |
| [16] |
H. Gao, J. Chen, Y. Zhang, et al., Chem. Eng. J. 306 (2016) 522-530. DOI:10.1016/j.cej.2016.07.080 |
| [17] |
Y. Qian, G. Xue, J. Chen, et al., J. Hazard. Mater. 354 (2018) 153-160. DOI:10.1016/j.jhazmat.2018.05.004 |
| [18] |
C. Tan, N. Gao, Y. Deng, et al., Sep. Purif. Technol. 109 (2013) 122-128. DOI:10.1016/j.seppur.2013.03.003 |
| [19] |
C. Zhu, F. Zhu, C. Liu, et al., Environ. Sci. Tech. 52 (2018) 8548-8557. DOI:10.1021/acs.est.7b06279 |
| [20] |
O.S. Furman, A.L. Teel, R.J. Watts, Environ. Sci. Technol. 44 (2010) 6423-6428. DOI:10.1021/es1013714 |
| [21] |
G.V. Buxton, C.L. Greenstock, W.P. Helman, A.B. Ross, J. Phys. Chem. 17 (1988) 513-886. DOI:10.1063/1.555805 |
| [22] |
L. Zhou, C. Ferronato, J. Chovelon, Chem. Eng. J. 311 (2017) 28-36. DOI:10.1016/j.cej.2016.11.066 |
| [23] |
R. Zhang, P. Sun, T.H. Boyer, et al., Environ. Sci. Technol. 49 (2015) 3056-3066. DOI:10.1021/es504799n |
| [24] |
K. Hildenbrand, G. Behrens, D. Schulte-Frohlinde, et al., J. Chem. Soc. Perkin Trans. 2 (1989) 283-289. |
| [25] |
X. Luo, Z. Zheng, J. Greaves, et al., Water Res. 46 (2012) 1327-1336. DOI:10.1016/j.watres.2011.12.052 |
| [26] |
L. Zhou, Q. Zhao, X. Yang, et al., Chem. Eng. J. 394 (2020) 124839. DOI:10.1016/j.cej.2020.124839 |
| [27] |
P. Gara, G. Bosio, M. Gonzalez, et al., Photochem. Photobiol. Sci. 8 (2009) 992-997. DOI:10.1039/b900961b |