 2021, Vol. 32
2021, Vol. 32 


b School of Mathematics and Physics, Nanyang Institute of Technology, Nanyang 473004, China;
c College of Physics and Electronic Engineering, Nanyang Normal University, Nanyang 473061, China;
d Shaanxi Key Laboratory of Optoelectronic Functional Materials and Devices, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China;
e College of Resources and Environmental Engineering, Tianshui Normal University, Tianshui 741001, China;
f School of Sciences, Xi'an Technological University, Xi'an 710021, China
The successful exfoliation of graphene in 2004 opens the door of two-dimensional (2D) materials [1]. Different from their bulk materials, 2D materials possess unique optical, electronic, and mechanical properties, which show great potential in applications in next-generation electronic, optoelectronic, spintronic, and other novel devices. Take graphene as an example, the unique 2D honeycomb structure together with the sp2 hybridized C-C covalent bonds endows graphene with some special chemical and physical properties. Firstly, graphene possesses very high carrier mobility (~1.5 × 104 cm2 V−1 s−1) at room temperature. Secondly, graphene exhibits an excellent mechanical property. Thirdly, graphene shows very high thermal conductivity (~5300 W/mK) [2]. However, the zero-gap of graphene is an obstacle to switch current on and off in field-effect transistors. So more other two-dimensional materials, such as h-BN [3, 4], BC3 [5, 6] and NC3 [7, 8], have been investigated theoretically and synthesized experimentally.
In theory, since both boron and nitrogen atoms also can form strong sp2 hybridized covalent bonds, a simple and effective method of designing new 2D materials is replacing partially or wholly the carbon atoms of graphene with boron or nitrogen atoms. The 2D materials designed by this method have both the honeycomb structure and sp2 hybridized covalent bonds the same as graphene, which are also called graphene-like 2D materials. Interestingly, the resulting graphene-like 2D materials can gain many fascinating properties such as a more suitable band gap, superconductivity, high carrier mobility, and high thermal conductivity. For example, the graphene-like h-BN [3, 4] with a band gap of 5.8 eV is called white graphene, which can be used as a substrate or gate dielectric layer to improve the performance of electronic/photonic devices based on graphene or other 2D materials. Graphene-like BC3 nanosheet has been theoretically suggested as a promising candidate for gas-sensitive materials [9]. The extraordinary property makes single-layer BC3 (SLBC) adaptable to many applications [10-13]. Graphene-like carbon-nitrogen materials C2N and C3N were fabricated by Mahmood et al. in 2015 [14] and in 2016 [15] respectively, which were also found to be semiconductors. More complicated ternary 2D BxCyNz materials also have been synthesized by using pyrolysis [16], chemical vapor deposition [17], and doping [18]. Some of the 2D BxCyNz still remain the honeycomb structure. For example, hexagonal graphitic BCN (h-BCN) [19] with one-atom thickness have been successfully synthesized. First-principles calculations on h-BCN predicted a direct electronic band gap of 1.5 eV [20] that is intermediate between gapless graphene and insulting h-BN. BC2N was theoretically predicted by using first-principles calculations [21] and success-fully synthesized by the chemical vapor deposition method in the experiment [22]. B4CN3 and B3CN4 were proposed theoretically to be potential spintronic materials [23]. Graphene-like BC6N single-layer is a semiconductor with a direct band gap of 1.3 eV [24]. So we can see that the 2D ternary B–C-N materials can effectively expand the graphene-like 2D material family, gaining new 2D materials with unexpected properties.
In addition to some significant applications mentioned above, 2D boron-carbon-nitride materials also have a wide range of applications in other fields, such as energy storage [25-29], solar cells [30], and biotechnologies [31]. More to the point, because 2D boron-carbon-nitride materials have a large surface area, which is conducive to gas adsorption, they may be potential catalytic materials [32-44]. As an important industrial raw material, CO2 can be electroreduced to value-added chemicals or fuels driven by low-grade renewable electricity, enabling both renewable energy storage and negative carbon cycle [45-51]. However, our research shows that the interaction between the pristine BC3N2 and CO2 is rather weak, which limits its immediate potential for application as a catalyst. What is exciting is that decorating foreign atoms is an effective way to tune the interaction between 2D materials and molecules [52-56]. Mo is cheaper than other precious metals, such as Ru, Rh, Pd, and Au, moreover, there are many studies on Mo loaded on 2D materials recently [57-61]. It is worth noting that Cui and co-workers reported that a single-Mo doped BN monolayer can be used as a promising catalyst for CO2 reduction to CH4 [62]. Because of this, we screened possible single-atom catalysts involving five transition metals (TM = Mo, Ru, Rh, Pd, and Au) anchored on the BC3N2 monolayer. The results show that a single-Mo-atom-loaded BC3N2 monolayer possesses excellent performance for activating CO2 molecules.
In this paper, we employed the automatic structure search algorithm to predict the possible stable structures of BC3N2. We identified an unusual stoichiometry, a BC3N2 monolayer, with a graphene-like structure, which meets the thermodynamical, dynamical, and mechanical stability requirements. Although all B, C, and N elements are non-metallic elements, the BC3N2 shows a metallic character. It was discussed in detail for the structural characteristics, stabilities, electronic structure, mechanical properties, and the adsorption properties of CO2 on BC3N2 and Mo/BC3N2 sheets.
The global minimum structure of BC3N2 monolayer was predicted using the particle-swarm optimization (PSO) method as implemented in the CALYPSO code [63, 64]. Unit cells containing 1–4 formula units (f.u.) were considered. The population size and the number of generations were both set to 30, which had been tested to give convergent results. All of the initial structures were fully relaxed including the atomic positions and lattice parameters. The structural optimizations and the electronic property calculations were carried out using density functional theory in the Vienna ab initio simulation package (VASP) [65]. The projector augmented-wave (PAW) pseudopotential was used to represent electron-ion interactions [66, 67]. The electron exchange-correlation energy was treated within the generalized gradient approximation (GGA), using the functional of Perdew, Burke, and Ernzerhof (PBE) [68]. The energy cutoff of the plane wave was set to be 520 eV and the Brillouin zone was sampled with a 9 × 9 × 1 Γ-centered Monkhorst-Pack k-point grid. The convergence was chosen as 10−5 eV and 2 × 10-2 eV/Å for energy and force, respectively. The B 2s22p1, C 2s22p2, N 2s22p3, O 2s22p4 and Mo 4s24p65s14d5 electron configurations were treated as valence electrons.
The kinetic stability of the BC3N2 monolayer was assessed by phonon dispersion calculations, with the finite displacement method implemented in the PHONOPY package [69]. To evaluate thermal stability, we performed ab initio molecular dynamics (AIMD) calculations using PBE function and PAW pseudopotential. AIMD simulation in the canonical ensemble (NVT) lasts for 15 ps with a 2 × 2 × 1 supercell. The temperature was controlled by using the Nose-Hoover method.
In light of the electronic structure of the molybdenum atom, the spin polarization was taken into account to attain the ground state energy, we chose the 3 × 3 × 1 supercell as a basic unit. A plane wave cutoff energy for 400 eV was used. The Brillouin zone was sampled with a 3 × 3 × 1 Γ-centered Monkhorst-Pack k-point grid, and a Γ-centered Monkhorst-Pack grid of 5 × 5 × 1 was used for the final density of states (DOS) calculations. 10−5 eV and 2 × 10−2 eV/Å were chosen as the convergence accuracies of energy and force, respectively. A large vacuum space of 20 Å in the perpendicular direction of the sheet was used to avoid the interactions between periodic images. The van der Waals correction was also estimated employing the DFT-D2 method [70].
The adsorption energy (Eads) of an atom or a molecule (A: Mo or CO2) on a substrate (B: BC3N2 or Mo/BC3N2) was calculated using the expression: Eads = EAB − EA − EB, where EA(B) represents the total energy of A (or B) and EAB represents the total energy of the system with an adsorbate. According to the above definition, negative adsorption energy suggests that the adsorption process is exothermic and the adsorption system is thermodynamically stable. Contrarily, a positive value corresponds to endothermic and unstable adsorption. Bader charge analysis [71] was used to evaluate the atomic charges and electron transfer in the chemical reactions. The difference charge density was calculated to gain insights into the bonding nature. The crystal orbital Hamiltonian population (COHP) formalism was used to analyze the bonding features of the Mo/BC3N2 and CO2 adsorbed on Mo/BC3N2, as implemented in the LOBSTER package [72-74].
The stable structure of BC3N2 obtained through the global structure searching is shown in Fig. 1a, from which we found that the 2D structure exhibits planar honeycomb lattice. Two dimensional BC3N2 possesses a space group of P-62m (No. 189) and one unit cell of BC3N2 monolayer consists of one B, three C, and two N atoms with optimized lattice constants of a = b = 4.275 Å. Each of the B atom and N atom binds to three C atoms and the bond lengths of B-C and C-N are 1.484 and 1.397 Å, respectively, indicating that sp2 hybridized B-C and C-N bonds exist in BC3N2. The chemical bonding of the BC3N2 can be understood from its charge difference density (Fig. 1b), which is defined as the total electronic density of the BC3N2 minus the electron density of isolated B, C, and N atoms at their respective positions. It is obviously seen from Fig. 1b that the atoms between B-C and C-N are covalently bonded. The calculated band structure, as well as its projected density of states, are shown in Fig. 1c, different from the graphene and h-BN monolayer, the BC3N2 plane is metallic with a large density-of-states (DOS) at the Fermi level. The metallic character in BC3N2 is due to the contribution of 2p states of B, C and N atoms to the DOS at the Fermi level.

|
Download:
|
| Fig. 1. (a) Schematic illustration of two dimensional BC3N2. B, C and N atoms are represented by green, brown, and gray spheres, respectively. (b) Difference charge density of BC3N2. The gold color (i.e., 0.02 eÅ−3) in the map shows an increased electron density after bonding. (c) Electronic band structure calculated by using the PBE functional with the projected density of states (states per eV per unit cell) of BC3N2. (d) Phonon dispersion curves of BC3N2 monolayer. | |
{kind=link}
To clarify the stability of predicted two dimensional BC3N2, we estimated its formation energy defined as Ef = EBC3N2 ‒ EBC3 ‒ EN2, where EBC3N2, EBC3 and EN2 are the total energies of BC3N2 monolayer, BC3 monolayer, and the nitrogen, respectively. The calculated formation energy for the BC3N2 monolayer is ‒0.08 eV per f.u. The negative formation energy indicates the stability of the BC3N2 plane, and also implies that the synthesis of the BC3N2 under ambient conditions is feasible. The dynamical stability of BC3N2 was assessed by calculating the phonon dispersion curves. As shown in Fig. 1d, no imaginary frequency in the first Brillouin zone is found, which confirms the dynamical stability of BC3N2. Then, we performed AIMD simulations of the BC3N2 with a 2 × 2 × 1 supercell at the temperature of 2400 K and 2700 K, which can provide a fundamental evaluation in the thermodynamic stability of the given 2D material [75-79]. The fluctuation of the total potential energy with simulation time at 2400 K is plotted in Fig. S1 (Supporting information), which shows that the average value of the total potential energy remains nearly constant during the entire simulation. From the snapshot taken at the end of 15 ps, one can see that the original geometry of BC3N2 is generally well-kept and no bond is broken at 2400 K. The above results reveal that the BC3N2 monolayer can maintain its structural integrity at a high temperature of 2400 K.
The mechanical properties were investigated by calculating its linear elastic constants using the formulas [80], which have been given in Table S1 (Supporting information). Due to the symmetry, the hexagonal structures have C11 = C22 and an additional relation which is C66 = 0.5(C11 ‒ C12). The four independent elastic constants (C11, C22, C12, C66) of BC3N2 monolayer were calculated to be C11 = C22 = 329.4 N/m, C12 = 59.2 N/m, and C66 = 135.1 N/m, respectively. All the calculated elastic constants meet the necessary mechanical equilibrium conditions [81] for mechanical stability: C11C22 ‒ C122 > 0, and C 11, C22, C66 > 0, indicating that BC 3N2 monolayer is a favorable structure from the mechanical point of view. According to the elastic constants, Young's modulus (in-plane stiffness) for strains in the Cartesian directions is E = (C11C22 ‒ C122)C11−1. For BC3N2, as shown in Table S1, the in-plane stiffness is calculated to be 319.0 N/m, which is lower than that of graphene (350.0 N/m) [82] and much higher than other reported values such as MoS2 monolayer (125.0 N/m) [83], silicene (71.2 N/m) [84], and germanene (42.0 N/m) [85].
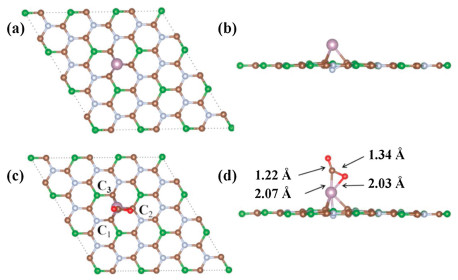
We first studied the adsorption energies of CO2 molecule on the BC3N2 surface at the possible sites, including the hollow site of B-C-N (H), the top sites of B(T1), C(T2), and N(T3) atoms, and the bridge sites of B-C (Br1) and N-C bonds (Br2). Two adsorption manners were designed, namely, the CO2 molecule being parallel and vertical to the BC3N2 plane. Exact adsorption sites are hard to fathom for the actual adsorption process, but these selected sites are highly representative and capable to simulate all the possible adsorption situations. As shown in Table S2 (Supporting information), due to the sp2 hybridized B-C and C-N bonds in the BC3N2 sheet, CO2 is difficult to adsorb on the surface of BC3N2. Therefore, we decorated the atoms on BC3N2 for changing the electronic structure of the pristine monolayer. As shown in Table S3 (Supporting information), compared with the other noble metal atoms, such as Ru, Rh, Pd and Au, Mo atoms can bond with BC3N2 sheet better. Therefore, it was chosen to functionalize the BC3N2 surface. To determine the most stable configuration for the Mo atom on the BC3N2 surface, we performed a scan of the energy of the adsorbed atom at the six adsorption sites mentioned above on the BC3N2 sheet. From the energy point of view, the Mo atom prefers to be adsorbed at the T3 site with an Eads of ‒3.49 eV, which is lower than those at the other sites in Table S2, showing that the Mo/BC3N2 system is relatively stable in thermodynamics. As shown in Fig. 2a and b, the stable structure of Mo/BC3N2 is formed from one Mo atom connected with three surrounding C atoms and the Mo atom is just above the center of an equilateral triangle consisted of three carbon atoms. Because of the strong interactions between Mo and C (C1, C2, and C3) atoms, the latter protrudes outside from the BC3N2 substrate with the N atom heavily sinking. As is seen from Table S4 (Supporting information), each of the three C atoms is equidistant from Mo, and the distance is nearly 2.16 Å, which is 2.34 Å for Mo and N atoms. Then we discussed the interaction between the CO2 molecule and Mo/BC3N2 monolayer. The Possible initial configurations were optimized to find out the most favorable geometrical structure. After strict calculations, the optimal structure of CO2 adsorption on Mo/BC3N2 system is given in Figs. 2c and d, we can see that the Ob and C atoms of CO2 molecule bind to Mo which is loaded on BC3N2 sheet. In contrast to Mo/BC3N2, the distances between Mo and three C (C1, C2 and C3) atoms change obviously. As given in Table S4, the C1 atom is closer to the Mo atom, the distance between them has been shortened from 2.16 Å to 2.11 Å, which decreases by about 2.3% comparing with the previous value, and the distance is also getting shorter between the C3 and Mo atoms, which decreases from 2.16 Å to 2.12 Å, however, it increases from 2.16 Å to 2.21 Å for the distance between the C2 atom and Mo atom. Finally, we needed to pay attention to the distance given in Table S4 between N and Mo atoms, which decreases from 2.34 Å to 2.20 Å. The changes mentioned above are mainly because of the adsorption of CO2 molecules. The interaction is mutual so that the geometric configuration of the adsorbed CO2 molecule must have changed accordingly. In terms of bond lengths shown in Table S4, the C-Oa bond length is elongated from 1.17 Å to 1.22 Å around the interaction area, and in particular, we should note that it significantly increases from 1.17 Å to 1.34 Å for the distance between C and Ob. Moreover, the CO2 molecule also features an Oa-C-Ob bond angle of 132.23 degrees, which also changes obviously. The results indicate that the CO2 molecule is activated. Through the above analysis, we can know that compared with the pristine BC3N2 surface, Mo/BC3N2 has a stronger adsorption affinity to CO2 molecules. The variation probably has something to do with charge redistribution and the bonding nature, which will be discussed in the following sections in terms of the Partial density of states (PDOS), COHP, and Bader charge

|
Download:
|
| Fig. 2. Top and side views of the optimized geometric structures of (a, b) Mo/BC3N2 and (c, d) CO2 adsorbed on Mo/BC3N2. | |
{kind=link}
Firstly, we analyzed the electronic structure of Mo/BC3N2. Fig. 3a shows the partial DOS (PDOS) map for the BC3N2 system with Mo adsorption. The Mo 4d state crosses the Fermi level, indicating that Mo/BC3N2 also possesses metallic conductivity. The energy states around the Fermi level are mainly from the Mo 4d and N 2p states, suggesting that the Mo 4d and N 2p states dominate the conductivity of Mo/BC3N2. Furthermore, the density of states near the Fermi level is obviously asymmetric, thereby the Mo/BC3N2 system has a magnetic character. The PDOS of the BC3N2 system change greatly nearby the Fermi level when loaded with Mo, and the PDOS peaks for the Mo 4d state are wider and higher than those of the other atomic states of Mo/BC3N2, which implies the high activity of Mo/BC3N2 and also indicates a strong interaction between the Mo atom and BC3N2 sheet. Thus, loading with the Mo atom is responsible for the electronic and magnetic properties of the Mo/BC3N2 system. Besides, as shown in Table S4, an apparent charge of 0.66e is transferred from Mo to the BC3N2 sheet, which further demonstrates that Mo is chemisorbed on the BC3N2 monolayer. In Fig. 3b, we can see that the Mo atom interacts with the BC3N2 through ionic bonds with electrons (yellow) gathering on the surface of the BC3N2. The existence of ionic bonds between Mo and C (C1, C2 and C3) atoms can be attributed to the significant difference in electronegativity. In addition, as depicted in Fig. S2 (Supporting information), there is a very weak bonding between Mo and N, but the bonds between three C atoms and Mo are much stronger to make the system stable. Secondly, we considered the adsorption of CO2 on Mo/BC3N2. Combined with the above geometric configuration analysis of CO2 absorbed on Mo/BC3N2, it can be seen that, due to the adsorption of CO2 on Mo/BC3N2, there is a profound impact on the electronic structure of the Mo/BC3N2. The PDOS map, as depicted in Fig. 3c, shows that the adsorption of CO2 does not change the fact that the system is magnetic and metallic. But comparing with the Mo/BC3N2 sheet, the PDOS peaks change obviously for the Mo 4d states when adsorbed with CO2, indicating that the Mo atom's contribution is no longer overwhelming to the total density of states around the Fermi level. In Figs. 4a and b, it can be seen that both Ob and C atoms bond with Mo. In Fig. 3d, the differential charge density analysis further illustrates that the ionic feature exists in the Mo-C and Mo-Ob. As shown in Figs. 4c-e and Figs. S2a-c, before and after CO2 adsorption, there are slight changes for the interactions between Mo and C (C1, C2, and C3) atoms in the BC3N2 substrate. Most notably, by comparing Fig. 4f and Fig. S2d, we found that it is significantly enhanced for the bonding between Mo and N atoms. Meanwhile, it has changed significantly for the bonding between C and each of the two oxygen atoms, that is, the C-O bonds become weaker, which is easily found in Figs. 4g–i. This is consistent with the results of the geometric configuration analysis mentioned above. According to the analysis of the Bader charge, it gains 0.9 e for CO2 molecule from Mo/BC3N2 system, which also explains why the bonding between C and O is weaker. Finally, it should be emphasized that Mo decorated on BC3N2 support can significantly change the electronic structure of the BC3N2 sheet. Moreover, it is also changed for the adsorption behavior as well as the molecular structure of CO2 loaded on Mo/BC3N2, namely, the ability to capture CO2 for Mo/BC3N2 is far better than that of the pristine BC3N2, and the charges (0.9 e) given in Table S4 transfers from the Mo atom to the CO2 molecule, facilitating the activation of CO2 on Mo/BC3N2.

|
Download:
|
| Fig. 3. Partial DOS (PDOS) maps for (a) Mo/BC3N2 and (c) CO2 adsorbed on Mo/BC3N2. Difference charge density of (b) Mo/BC3N2 and (d) CO2 adsorbed on Mo/BC3N2. The yellow and green isosurfaces with an isovalue of 0.03 eÅ−3 correspond to charge accumulation and depletion, respectively. | |
{kind=link}

|
Download:
|
| Fig. 4. The COHP of Mo (or O)-C (or N) pair(s) of the CO2 adsorbed on Mo/BC3N2. (a) Mo-Ob, (b)Mo-C, (c) Mo-C1, (d)Mo-C2, (e)Mo-C3, (f)Mo-N, (g) C-Ob, (h) C-Oa and (i) the COHP of C-O pairs of the gas CO2. The Fermi level is at the energy origin electronegativity. | |
{kind=link}
In conclusion, we search a new metallic BC3N2 monolayer by using the PSO-based global structure search method and first-principles calculations, which is stable thermodynamically, dynamically, and mechanically. Ab initio molecular dynamics (AIMD) simulation shows that BC3N2 system has thermal stability. Moreover, the BC3N2 has an in-plane stiffness comparable to that of graphene. The study on CO2 adsorption shows that the interaction between the pristine BC3N2 and CO2 is rather weak due to the sp2 hybridized B-C and C-N bonds in the BC3N2 sheet. However, the decoration of the Mo atom greatly enhances the interaction between gas molecule CO2 and Mo/BC3N2, which demonstrates that metallic atom decorated on BC3N2 monolayer can change the adsorption capacity of BC3N2 to a great extent towards gas molecule CO2, facilitating the activation of CO2 on Mo/BC3N2. According to our research, it can be predicted that metal-modified BC3N2 may be an excellent catalyst if BC3N2 is loaded with an appropriate metallic atom.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (Nos. 21603109, U1404216, U1904179, U1404608), the Special Fund of Tianshui Normal University, China (Grant No. CXJ2020-08), the Key Science Fund of Educational Department of Henan Province of China (Nos. 19A140013, 20B140010) and Shaanxi Provincial Education Department Serves Local Scientific Research Program (Nos. 19JC020, 20JK0676).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.046.
| [1] |
K.S. Novoselov, A.K. Geim, S.V. Morozov, et al., Science 306 (2004) 666-669. DOI:10.1126/science.1102896 |
| [2] |
A.A. Balandin, S. Ghosh, W. Bao, et al., Nano Lett. 8 (2008) 902-907. DOI:10.1021/nl0731872 |
| [3] |
I. Jo, M.T. Pettes, J. Kim, et al., Nano Lett. 13 (2013) 550-554. DOI:10.1021/nl304060g |
| [4] |
K. Watanabe, T. Taniguchi, H. Kanda, Nat. Mater. 3 (2004) 404-409. DOI:10.1038/nmat1134 |
| [5] |
H. Tanaka, Y. Kawamataa, H. Simizua, et al., Solid State Commun. 136 (2005) 22-25. DOI:10.1016/j.ssc.2005.06.025 |
| [6] |
Q. Wang, L.Q. Chen, J.F. Annett, Phys. Rev. B. 54 (1996) R2271. DOI:10.1103/PhysRevB.54.R2271 |
| [7] |
Q. Hu, Q. Wu, H. Wang, et al., Phys. Status Solidi B 249 (2012) 784-788. DOI:10.1002/pssb.201147319 |
| [8] |
C.H. Lee, D. Zhang, Y.K. Yap, J. Phys. Chem. C 116 (2012) 1798-1804. DOI:10.1021/jp2112999 |
| [9] |
S.M. Aghaei, M.M. Monshi, I. Torres, et al., Appl. Surf. Sci. 427 (2018) 326-333. DOI:10.1016/j.apsusc.2017.08.048 |
| [10] |
R.P. Joshi, B. Ozdemir, V. Barone, et al., J. Phys. Chem. Lett. 6 (2015) 2728-2732. DOI:10.1021/acs.jpclett.5b01110 |
| [11] |
B. Mortazavi, M. Shahrokhi, M. Raeisi, et al., Carbon 149 (2019) 733-742. DOI:10.1016/j.carbon.2019.04.084 |
| [12] |
S.M. Aghaei, M.M. Monshi, I. Torres, et al., Appl. Surf. Sci. 427 (2018) 326-333. DOI:10.1016/j.apsusc.2017.08.048 |
| [13] |
N. Ashraf, A. Majid, M. Rafique, et al., M. B. Chin. J. Phys. 66 (2020) 246-257. DOI:10.1016/j.cjph.2020.03.035 |
| [14] |
J. Mahmood, E.K. Lee, M. Jung, et al., Nat. Commun. 6 (2015) 1-7. |
| [15] |
J. Mahmood, E.K. Lee, M. Jung, et al., Proc. Natl. Acad. Sci. 113 (2016) 7414-7419. DOI:10.1073/pnas.1605318113 |
| [16] |
M.A. Mannan, H. Noguchi, T. Kida, et al., Thin Solid Films 518 (2010) 4163-4169. DOI:10.1016/j.tsf.2009.11.086 |
| [17] |
D. Kurapov, D. Neuschütz, R. Cremer, et al., Vacuum 68 (2002) 335-339. DOI:10.1016/S0042-207X(02)00506-7 |
| [18] |
C.N.R. Rao, K. Gopalakrishnan, A. Govindaraj, Nano Today 9 (2014) 324-343. DOI:10.1016/j.nantod.2014.04.010 |
| [19] |
D.P. Yang, Y.A. Li, X.X. Yang, et al., Chin. Phys. Lett.. 24 (2007) 1088. DOI:10.1088/0256-307X/24/4/066 |
| [20] |
S. Beniwal, J. Hooper, D.P. Miller, et al., ACS Nano 11 (2017) 2486-2493. DOI:10.1021/acsnano.6b08136 |
| [21] |
A.Y. Liu, R.M. Wentzcovitch, M.L. Cohen, Phys. Rev. B 39 (1989) 1760-1765. |
| [22] |
H.A. Castillo, P.J. Arango, J.M. Vélez, et al., Surf. Coat. Technol. 204 (2010) 4051-4056. DOI:10.1016/j.surfcoat.2010.05.025 |
| [23] |
H. Pan, Y. Sun, Y. Zheng, et al., New J. Phy. 18 (2016) 093021. DOI:10.1088/1367-2630/18/9/093021 |
| [24] |
A. Bafekry, Physica E. 118 (2020) 113850. DOI:10.1016/j.physe.2019.113850 |
| [25] |
S. Ahmad, X. Guo, Chin. Chem. Lett. 29 (2018) 657-663. DOI:10.1016/j.cclet.2017.08.057 |
| [26] |
B. Xu, S. Qi, M. Jin, et al., Chin. Chem. Lett. 30 (2019) 2053-2064. DOI:10.1016/j.cclet.2019.10.028 |
| [27] |
D. Zhou, C. Li, F. Yin, et al., Chin. Chem. Lett. 31 (2020) 2325-2329. DOI:10.1016/j.cclet.2020.04.045 |
| [28] |
C. Pu, J. Yu, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 1081-1085. DOI:10.1016/j.cclet.2020.08.042 |
| [29] |
Y. Song, X. Li, C. He, Chin. Chem. Lett. 32 (2021) 1106-1110. DOI:10.1016/j.cclet.2020.08.024 |
| [30] |
X. Wang, L. Zhi, K. Müllen, Nano Lett. 8 (2008) 323-327. DOI:10.1021/nl072838r |
| [31] |
S. He, B. Song, D. Li, et al., Adv. Funct. Mater. 20 (2010) 453-459. DOI:10.1002/adfm.200901639 |
| [32] |
X. Zhang, A. Chen, Z. Zhang, et al., J. Mater. Chem. A 6 (2018) 11446-11452. DOI:10.1039/C8TA03302A |
| [33] |
Y. Zhou, G. Gao, J. Kang, et al., J. Mater. Chem. A 7 (2019) 12050-12059. DOI:10.1039/C9TA01389J |
| [34] |
F. Yu, L. Wang, Q. Xing, et al., Chin. Chem. Lett. 31 (2020) 1648-1653. DOI:10.1016/j.cclet.2019.08.020 |
| [35] |
W. Wu, Z. Ao, T. Wang, et al., Phys. Chem. Chem. Phys. 16 (2014) 16588-16594. DOI:10.1039/C4CP01416B |
| [36] |
G. Liu, J. Zhou, W. Zhao, et al., Chin. Chem. Lett. 31 (2020) 1966-1969. DOI:10.1016/j.cclet.2019.12.023 |
| [37] |
Q.G. Jiang, Z.M. Ao, S. Li, et al., RSC Adv. 4 (2014) 20290-20296. DOI:10.1039/C4RA01908C |
| [38] |
Y. Chen, S. Lan, M. Zhu, Chin. Chem. Lett. 32 (2021) 2052-2056. DOI:10.1016/j.cclet.2020.11.016 |
| [39] |
X. Wang, H. Gao, C. Zhai, et al., Ind. Eng. Chem. Res. 59 (2020) 19252-19259. DOI:10.1021/acs.iecr.0c03436 |
| [40] |
J. Hu, C. Zhai, H. Gao, et al., Sustain. Energy Fuels 3 (2019) 439-449. DOI:10.1039/C8SE00507A |
| [41] |
S. Hu, Y. Yu, Y. Guan, et al., Chin. Chem. Lett. 31 (2020) 2839-2842. DOI:10.1016/j.cclet.2020.08.021 |
| [42] |
H. Ji, P. Du, D. Zhao, et al., Appl. Catal. B: Environ.. 263 (2020) 118357. DOI:10.1016/j.apcatb.2019.118357 |
| [43] |
C.C. Wang, X. Wang, W. Liu, Chem. Eng. J. 391 (2020) 123601. DOI:10.1016/j.cej.2019.123601 |
| [44] |
L. Chen, H. Ji, J. Qi, et al., Chem. Eng. J.. 406 (2021) 126877. DOI:10.1016/j.cej.2020.126877 |
| [45] |
M. Asadi, K. Kim, C. Liu, et al., Science 353 (2016) 467-470. DOI:10.1126/science.aaf4767 |
| [46] |
M. Liu, Y. Pang, B. Zhang, et al., Nature 537 (2016) 382-386. DOI:10.1038/nature19060 |
| [47] |
L. Zhang, Z.J. Zhao, J.L. Gong, Angew. Chem. 129 (2017) 11482-11511. DOI:10.1002/ange.201612214 |
| [48] |
L. Wang, L. Wang, J. Zhang, et al., Angew. Chem. Int. Ed 57 (2018) 6104-6108. DOI:10.1002/anie.201800729 |
| [49] |
S. Lee, G. Park, J. Lee, ACS Catal. 7 (2017) 8594-8604. DOI:10.1021/acscatal.7b02822 |
| [50] |
J. Huo, L. Fu, C. Zhao, et al., Chin. Chem. Lett. 32 (2021) 2269-2273. DOI:10.1016/j.cclet.2020.12.059 |
| [51] |
L. Fu, R. Wang, C. Zhao, et al., Chem. Eng. J.. 414 (2021) 128857. DOI:10.1016/j.cej.2021.128857 |
| [52] |
H.K. Lee, Solid State Commun. 150 (2010) 1959-1962. DOI:10.1016/j.ssc.2010.08.028 |
| [53] |
O. Cabria, M.J. Lopez, J.A. Alonso, J. Chem. Phys. 123 (2005) 204721. DOI:10.1063/1.2125727 |
| [54] |
Y. Tang, D. Ma, W. Chen, et al., Sens. Actuators B: Chem. 211 (2015) 227-234. DOI:10.1016/j.snb.2015.01.057 |
| [55] |
C. He, R. Wang, H. Yang, et al., Appl. Surf. Sci.. 507 (2020) 145076. DOI:10.1016/j.apsusc.2019.145076 |
| [56] |
C. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci.. 509 (2020) 145392. DOI:10.1016/j.apsusc.2020.145392 |
| [57] |
P. Tan, Catal. Commun. 103 (2018) 101-104. DOI:10.1016/j.catcom.2017.10.008 |
| [58] |
S.J. Han, S.K. Kim, A. Hwang, et al., Appl. Catal. B: Environ. 241 (2019) 305-318. DOI:10.1016/j.apcatb.2018.09.042 |
| [59] |
M. Ameen, M.T. Azizan, A. Ramli, et al., Catal. Today 355 (2020) 51-64. DOI:10.1016/j.cattod.2019.03.028 |
| [60] |
J. Deng, J. Liu, W. Song, et al., RSC Adv. 7 (2017) 7130-7139. DOI:10.1039/C6RA27126J |
| [61] |
X. Gao, F. Zhang, Y. Yu, et al., Catal. Commun. 122 (2019) 47-51. DOI:10.1016/j.catcom.2018.12.019 |
| [62] |
Q. Cui, G. Qin, W. Wang, et al., Beilstein J. Nanotechnol. 10 (2019) 540-548. DOI:10.3762/bjnano.10.55 |
| [63] |
Y. Wang, J. Lv, L. Zhu, et al., Comput. Phys. Commun. 183 (2012) 2063-2070. DOI:10.1016/j.cpc.2012.05.008 |
| [64] |
Y. Wang, J. Lv, L. Zhu, et al., Phys. Rev. B Condens. Matter Mater. Phys.. 82 (2010) 094116. DOI:10.1103/PhysRevB.82.094116 |
| [65] |
G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [66] |
P.E. Blöchl, Phys. Rev. B 50 (1995) 17953-17979. |
| [67] |
G. Kresse, D. Joubert, Phys. Rev. B 59 (1999) 1758-1775. |
| [68] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865. DOI:10.1103/PhysRevLett.77.3865 |
| [69] |
A. Togo, F. Oba, I. Tanaka, Phys. Rev. B. 78 (2008) 134106. DOI:10.1103/PhysRevB.78.134106 |
| [70] |
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799. DOI:10.1002/jcc.20495 |
| [71] |
G. Henkelman, A. Arnaldsson, H. Jónsson, Comput. Mater. Sci. 36 (2006) 354-360. DOI:10.1016/j.commatsci.2005.04.010 |
| [72] |
R. Dronskowski, P.E. Bloechl, J. Phys. Chem. 97 (1993) 8617-8624. DOI:10.1021/j100135a014 |
| [73] |
V.L. Deringer, A.L. Tchougreeff, R. Dronskowsk, J. Phys. Chem. A 115 (2011) 5461-5466. DOI:10.1021/jp202489s |
| [74] |
S. Maintz, V.L. Deringer, A.L. Tchougreeff, et al., J. Comput. Chem. 37 (2016) 1030-1035. DOI:10.1002/jcc.24300 |
| [75] |
W. Xu, S. Ali, Y. Jin, et al., ACS Appl. Electron. Mater. 2 (2020) 3853-3858. DOI:10.1021/acsaelm.0c00686 |
| [76] |
N. Miao, J. Wang, Y. Gong, et al., Chem. Mater. 32 (2020) 6947-6957. DOI:10.1021/acs.chemmater.0c02139 |
| [77] |
B. Song, Y. Zhou, H. Yang, et al., J. Am. Chem. Soc. 141 (2019) 3630-3640. DOI:10.1021/jacs.8b13075 |
| [78] |
L. Wang, D. Wei, S. Kang, et al., J. Phys. Chem. C 122 (2018) 22911-22919. DOI:10.1021/acs.jpcc.8b05412 |
| [79] |
W. Xu, R. Wang, Y. Jin, et al., J. Phys. Chem. C 123 (2019) 16851-16856. DOI:10.1021/acs.jpcc.9b04219 |
| [80] |
R.C. Andrew, R.E. Mapasha, A.M. Ukpong, et al., Phys. Rev. B. 85 (2012) 125428. DOI:10.1103/PhysRevB.85.125428 |
| [81] |
F. Mouhat, F. Coudert, Phys. Rev. B. 90 (2014) 224104. DOI:10.1103/PhysRevB.90.224104 |
| [82] |
F. Liu, P. Ming, J. Li, Phys. Rev. B. 76 (2007) 064120. DOI:10.1103/PhysRevB.76.064120 |
| [83] |
Z. Zhang, Y. Yang, E.S. Penev, et al., Adv. Funct. Mater.. 27 (2017) 1605059. DOI:10.1002/adfm.201605059 |
| [84] |
Q. Peng, X. Wen, S. De, RSC Adv. 3 (2013) 13772-13781. DOI:10.1039/c3ra41347k |
| [85] |
R. John, B. Merlin, Cryst. Struct. Theory Appl. 5 (2016) 43-55. |