 2021, Vol. 32
2021, Vol. 32 


b College of Resources and Environmental Engineering, Tianshui Normal University, Tianshui 741001, China;
c Shaanxi Key Laboratory of Optoelectronic Functional Materials and Devices, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China;
d Institute of Environmental and Energy Catalysis, School of Materials Science and Chemical Engineering, Xi'an Technological University, Xi'an 710021, China;
e School of Sciences, Xi'an Technological University, Xi'an 710021, China;
f School of Environmental Science and Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China
Nitrogen (N2) is abundant, cheap, and readily available. Although the atmosphere contains more than 78% N2, it cannot be used or absorbed directly by humans, animals, or plants. N2 can be consumed only as nitrogen-containing compounds that can be obtained in food and by industrial production [1]. Ammonia (NH3) is an important nitrogen-containing compound, which is widely used in agriculture, pharmaceutical production, and other industrial processes. Currently, industrial N2 fixation for producing NH3 mainly relies on the Haber–Bosch process, which not only requires high energy consumption, but also generates a large number of greenhouse gases [2-5]. Therefore, the search for highly efficient and low-energy-consuming alternative catalysts has been a major focus area for researchers [6-16]. Over the years, several alternatives to the Haber–Bosch method have been developed, including chemical [6, 7], electrochemical [8-11] and photochemical routes [12-14].
Among other methods, electrochemical NRR has been extensively studied (ideally at room temperature) in recent years as a green technology for NH3 production. Compared with the traditional Haber–Bosch process, electrochemical NRR technology has many advantages, such as the use of renewable resources, moderate reaction conditions, and low equipment costs. This process is also fuelled by water rather than fossil fuels. However, due to the intrinsically inert properties of N2, its cleavage and reduction under ambient conditions presents several challenges. For example, (1) N2 has negative electron affinity (−1.8 eV) and high ionisation potential (15.85 eV), which can reduce its reactivity, (2) N≡N bond dissociation is difficult because of the high total bond energy (941 kJ/mol) and the thermodynamically high energy (410 kJ/mol) required for cleavage of the first bond, and (3) the large HOMO–LUMO energy gap (10.82 eV) is not favourable for electron transfer [17-19]. Therefore, it is necessary to use catalysts to weaken the bond energy of N2 and reduce the activation energy to achieve high efficiency and low energy consumption during NH3 synthesis. For this reason, the NRR process still poses great challenges for researchers and demands highly efficient electrocatalysts [20, 21]. The key factors governing the performance of electrochemical catalysts are as follows: (1) N2 adsorption and NH3 desorption on the catalyst; (2) the competitive relationship between the NRR and hydrogen evolution reaction (HER); and (3) the high equilibrium potential (−3.2 V in theory). Therefore, the design of novel and efficient electrocatalysts is a major technical challenge facing the scientific community. To date, many kinds of electrocatalysts for the NRR have been designed both theoretically and experimentally [22-47], especially transition metal (TM)-based catalysts [28-35, 40], which have been studied extensively in recent years.
It is well known that the chemisorption of N2 molecules onto the active centres, and subsequent activation with electrons transferred from the catalysts, are considered prerequisite steps for the NRR. The good reduction capability of TMs is commonly attributed to the combination of filled and empty d orbitals in their electronic structure. TM atoms have empty d orbitals to accept the lone-pair electrons from the N2 molecule, while simultaneously donating d electrons back to the antibonding π* orbitals of N2 to weaken the N≡N triple bond. Recently, various traditional TM electrocatalysts have been reported to be effective for the NRR. Many studies have reported that Fe is one of the most active TM catalysts for electrochemical synthesis of NH3. In 2017, Zhao et al. synthesized several metal-organic frameworks (MOFs) using a hydrothermal process. The prepared MOFs exhibited excellent catalytic activity for electrochemical NH3 synthesis at low temperatures and ambient pressure. Among these, the MOF (Fe) displayed the best catalytic activity; the highest NH3 formation rate and the highest current efficiency were 2.12 × 10−9 mol s−1 cm−2 and 1.43%, respectively, at 1.2 V and 90 ℃, when using pure N2 and H2O as raw materials [33]. Chen et al. studied Fe2O3 supported on carbon nanotubes (CNTs) as the catalyst, using which a rate of NH3 formation of 2.2 × 10−3 g m−2 h−1 was obtained at room temperature and atmospheric pressure with a flow of N2 gas and an applied potential of 2.0 V. Their results also indicated that the active sites in NH3 electrocatalytic synthesis may be associated with specific carbon sites formed at the interface between Fe particles and CNT. These were able to activate N2, making it more reactive towards hydrogenation [34]. Du et al. revealed that the NRR process can proceed preferentially with Fe in the presence of S, and the conversion of *NH2 to *NH3 was the potential-determining step, with an energy barrier of 0.45 eV according to density functional theory (DFT) calculations [35].
Interestingly, the non-metal B-based molecular catalyst also reacted favourably with N2. Notably, sp3-hybridised B also contains both occupied and empty orbitals, indicating its potential for N2 fixation. More importantly, sp3-hybridised B can bind N2 through a side-on pattern due to the compatibility of the orbitals [26, 27, 37-40]. For example, Ji et al. concluded that a boron-interstitial (Bint)-doped C2N layer can act as a metal-free electrocatalyst for N2 fixation and reduction to NH3 after performing extensive DFT computations. Their computations revealed that the Bint-doped C2N layer could sufficiently activate the N2 molecule via the "acceptance-donation" process due to its significant positive charge and the magnetic moment of the B dopant [26]. Liu et al. reported boron sheets doped with single Ru atoms exhibited outstanding catalytic activity for NH3 synthesis under ambient conditions via the distal reaction pathway, with a small activation barrier (0.42 eV) [27]. Qiu et al. fabricated a B4C nanosheet, which could be utilized as a superior metal-free electrocatalyst for the NRR with excellent selectivity at room temperature and ambient pressure [39]. These studies highlight the value of B as a substrate for the design of highly efficient electrocatalysts for the NRR. In summary, Fe is one of the most active metal catalysts and B forms a strong bond with N2. Therefore, Fe and B can be used in combination to form a new electrocatalyst, which can achieve high stability and high loading of active sites simultaneously. Despite the high N2 fixation capacity of Fe-B electrocatalysts, only a limited number have been reported. To our knowledge, only Li et al. have explored FeB, FeB2, FeB6 (alpha), and FeB6 (beta) as potential NRR catalysts under the framework of DFT. Their results revealed that FeB6 (beta) offered the best performance in terms of the lowest maximum energy required for the elementary steps (0.68 eV) [40]. Due to a gap in the literature regarding Fe-B electrocatalysts, we will design a new Fe-B electrocatalyst and study the NRR in the hope of achieving an improved catalytic effect.
In this study, we explore the potential of Fe2B2 as an NRR electrocatalyst using DFT. The theoretical calculations not only facilitate the exploration of efficient catalysts and predict trends in chemical reaction rates, but also provide an emerging understanding of the mechanisms and allow interpretation of experimental data with mechanistic insights and structure-reactivity correlations. Herein, our results demonstrate that the enzymatic mechanism is the most favourable NRR pathway of those tested, because it provides a lower limiting potential (−0.44 V), lower free energy (only 0.02 eV) of the first step hydrogenation reaction (*N–N to *NH–N), and more electron transfer from Fe2B2 to the reaction species. In addition, both vacancies and dopants can assist in reducing the energy barrier of the potential-determining step. This indicates that Fe-B catalysts have superior qualities which can enhance NRR via enzymatic pathways.
All the calculations are performed using spin-polarized DFT with the projected augmented wave (PAW) formalism, using the Vienna Ab initio Simulation Package (VASP) [48-51]. Electronic exchange and correlation effects are described by the generalized gradient approximation (GGA) as given by the Perdew, Burke, and Ernzerhof (PBE) equation. The cut-off energy for the plane-wave basis set is assumed to be 500 eV. The Brillouin-zone integration is conducted using a 5 × 5 × 1 Monkhorst-Pack grid. The convergence of the total energy is calculated to an accuracy of 10−5 eV and the Fermi smearing of the electronic levels is calculated using a width of 0.02 eV/Å. The Fe2B2 comprises a 2 × 2 supercell containing eight Fe and eight B atoms. A vacuum space exceeding 20 Å is employed to avoid interaction between two periodic units. The free NRR species are simulated using a unit cell with the dimensions 10 × 10 × 10 Å3. During the geometry optimisation, all atoms are allowed to relax.
The adsorption energy, Ead, which measures the stability of the adsorption configurations, is defined as (Eq. 1):

|
(1) |
where ET, EFe2B2 and Especies are the total energies of the adsorption system, Fe2B2 catalyst, and free NRR species, respectively. With this definition, a negative value indicates that the adsorption systems are energetically exothermic. The computational hydrogen electrode model [52] was used to calculate the free energy change (ΔG) in each of the reaction steps as follows (Eq. 2):
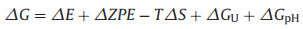
|
(2) |
where ΔE is obtained directly from DFT calculations, ΔZPE is the zero-point energy correction, and TΔS is the entropy change at room temperature (T = 298.15 K). ΔGU is the contribution of electrode potential (U) to ΔG. ΔGpH = kBT × ln10 × pH, where kB is the Boltzmann constant, T = 298.15 K and pH = 0 for the acid medium. The free energy of (H+ + e−) at standard conditions is assumed to be the energy of 1/2 H2 according to a computational hydrogen electrode model.
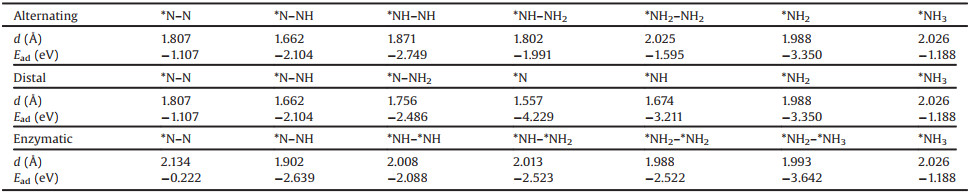
To catalyse the NRR, N2 molecules must be adsorbed on the catalyst surface; this is a prerequisite for initializing the NRR. The possible sites for adsorption of N2 on Fe2B2, include the top sites on Fe/B (TFe and TB), the hollow site on Fe/B (HFe and HB), and the bridge sites on Fe-B (TFe-TB for B1 and TFe-HB for B2) (Fig. 1a). Considering all forms of adsorption, including all end-on and side-on configurations, there are twelve adsorption sites. After optimisation, the most stable adsorption site is found to be the TFe site with end-on and side-on configurations (Fig. S1 in Supporting information). Through the adsorption energy calculation, we find that the end-on configuration (−1.107 eV) is more stable than the side-on configuration (−0.222 eV) with ΔEad = 0.885 eV. For both end-on and side-on configurations, the N-N bond distance after adsorption of N2 is elongated to 1.133 and 1.148 Å for end-on and side-on configurations, respectively, compared with 1.092 Å for free N2 gas, leading to the activation of the adsorbed N2 molecule. This happens because the charge transferred from Fe2B2 to N2 occupies the anti-bonding 2π* orbital of N2, lengthening the N-N bond. To gain more insight into the bonding nature and stability of Fe2B2, we plotted the partial density of states (PDOS), (Fig. 1b). The PDOS can not only reflect the distribution of electrons in each orbital, but also reveal the interaction between atoms and the information of chemical bonds. Fig. 1b demonstrates that the PDOS crosses the Fermi level, which indicates that Fe2B2 belongs to the metal system. In addition, there are two peaks on two sides of the Fermi level, and the relative width between the two peaks further demonstrates the high stability of Fe2B2 system. During the NRR, the high stability of Fe2B2 can also ensure an improved catalytic reaction.

|
Download:
|
| Fig. 1. (a) The top and side views for the possible adsorption sites of Fe2B2. The blue and pink balls represent Fe and B atoms, respectively. (b) The projected density of states (PDOS) of Fe2B2. | |
In general, there are three possible reaction pathways in the NRR process, the distal and alternating reaction pathways that involve end-on configurations and the enzymatic reaction pathway, which uses a side-on configuration (Fig. S2 in Supporting information). All possible adsorption structures of the various reaction species on Fe2B2 involved in the NRR mechanism have been studied; they include: *N2, *N=NH, *NH=NH, *NH–NH2, *NH2–NH2, *NH2–NH3, *N=NH2, *N, *NH, *NH2 and *NH3. The bond length and the calculated adsorption energy (Ead) for each species on Fe2B2 are listed in Table 1. In the following sections, we will describe these reaction pathways in detail.
|
|
Table 1 The values of d (in Å) is the bond length of Fe-N. The adsorption energy (eV) for each species on Fe2B2. |
{kind=link}
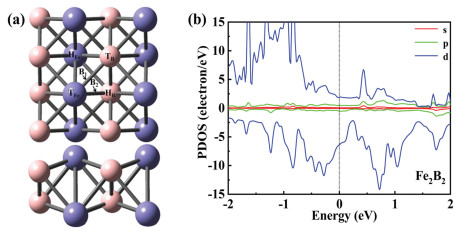
The electron pairs alternately attack two N atoms with the alternating mechanism. Our optimised structures of the reaction species are shown in Fig. 2a and the bond lengths are listed in Table 1. Generally, the bond length represents bonding stability. The more electrons participating in the bonding, the shorter the bond length is. Overall, the Fe-N bond length increased as the hydrogenation proceeded. However, there are exceptions with *NH=NH and *NH2, such as when *NH=NH reacts with the side-on configuration and NH2 adsorbs on the HFe site. Next, to evaluate the structural stability of various reaction species on Fe2B2, we have calculated the corresponding adsorption energy (Table 1). The adsorption energy reflects the strength of adsorption. Eq. 1 represents the positive correlation between the absolute value of adsorption energy and the strength of the interaction force, which is also an indicator of the stability of the system. As shown in Table 1, the adsorption energy is negative, and the absolute value is greater than 1 eV, which suggests that the reaction species are chemisorbed on Fe2B2 and have high stability. The NRR activity is further examined by Gibbs free energy profiles. Finally, we plotted the free energy (ΔG) (Fig. 2b). The highest energy barrier (ΔGmax), viz. the barrier for the potential-determining step (PDS), determines the thermodynamic feasibility of the electrocatalytic process. As depicted in Fig. 2b, despite the effective adsorption of N2 on Fe2B2 with a negative ΔG value (−0.61 eV), the first hydrogenation step is very difficult due to the weak N2H binding, requiring a high energy input (ΔG = 1.26 eV) for *N2 → *N2H (*N–N + H+ + e− = *N–NH). This is the PDS and the corresponding limiting potential is as high as −1.26 V.

|
Download:
|
| Fig. 2. (a) Optimized geometric structures of alternating mechanism for N2 reduction to NH3. (b) Free energy diagrams for the NRR on Fe2B2 through alternating mechanism at different applied potentials. | |
{kind=link}
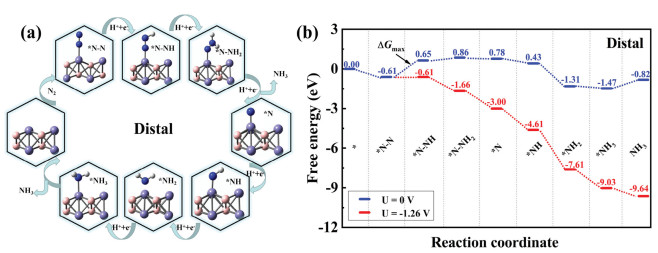
In contrast to the alternating mechanism, the electron pairs attack one N atom preferentially to produce NH3, followed by the other N atom in the case of the distal mechanism. Figs. 3a and b display the optimised structures of reaction species and the distal mechanism free energy diagrams. The corresponding bond lengths and adsorption energy are summarised in Table 1. The data for adsorption energy and bond length for the adsorption of various species on Fe2B2 demonstrates that the absolute value of adsorption energy is highest (−4.229 eV) for the N atom and the bond length is shortest for Fe-N (1.557 Å). In addition, we observed that the distal mechanism results in a higher adsorption energy and shorter bond length than the alternating mechanism for the steps where the two methods differ from one another. This indicates that the adsorption process is more stable when using the distal mechanism. We also analysed the whole hydrogenation process. The results reveal that the first step (*N–N + H+ + e− = *N–NH) and second step (*N–NH + H+ + e− = *N–NH2) are endothermic and the rest of the hydrogenation steps are exothermic. However, the first hydrogenation step with the free energy of 1.26 eV is still the PDS. Therefore, the efficiency of the NRR on Fe2B2 via the alternating or distal mechanism is very low.

|
Download:
|
| Fig. 3. (a) Optimized geometric structures of distal mechanism for N2 reduction to NH3. (b) Free energy diagrams for the NRR on Fe2B2 through distal mechanism at different applied potentials. | |
{kind=link}
In the enzymatic mechanism, N2 can adsorb on Fe2B2 through the side-on configuration, and subsequently two N atoms are hydrogenated alternately. The second NH3 molecule will be released immediately following the generation of the first one. From the change in adsorption energy and bond length (Table 1), we concluded that shorter bond length corresponds to stronger adsorption energy. Figs. 4a and b show the optimised structures of the reaction species and the corresponding free energy diagrams. Based on the calculated free energy of each elementary step, our results reveal that the hydrogenation of *N–*NH to *NH–*NH (*N–*NH + H+ + e− = *NH–*NH) is the PDS with the most positive free energy change (ΔG = 0.44 eV). Thus, the limiting potential of the whole process is −0.44 V, which is much lower than that for the alternating and distal mechanisms (−1.26 V). Thus, once an extra potential of U = −0.44 V is applied to eliminate the PDS barrier, the whole NRR process becomes energetically preferable. Although the configuration with the N2 adsorbed side-on is less stable than the end-on configuration, the free energy of the first hydrogenation step (*N–*N + H+ + e− = *N–*NH) is only 0.02 eV, which is far lower than the free energy values for the alternating and distal mechanism (1.26 eV). The subsequent steps are all exothermic. Previous DFT studies have revealed that the first hydrogenation step for generating or binding *N2H is the most important for the entire NRR process [53-55]. Therefore, the catalytic activity of the electrocatalyst depends on the strength of N2 adsorption and the difficulty of *N2H formation. This conclusion is consistent with those derived from previous results [26]. The above results reveal that the free energy for the Fe2B2 catalytic reaction via the enzymatic mechanism is lower than that of FeB6 (beta) (ΔG = 0.68 eV) [40] and close to that of Ru atom doped boron sheets (ΔG = 0.42 eV) [27], suggesting that Fe2B2 is a promising catalyst for the NRR.

|
Download:
|
| Fig. 4. (a) Optimized geometric structures of enzymatic mechanism for N2 reduction to NH3. (b) Free energy diagrams for the NRR on Fe2B2 through enzymatic mechanism at different applied potentials. | |
{kind=link}
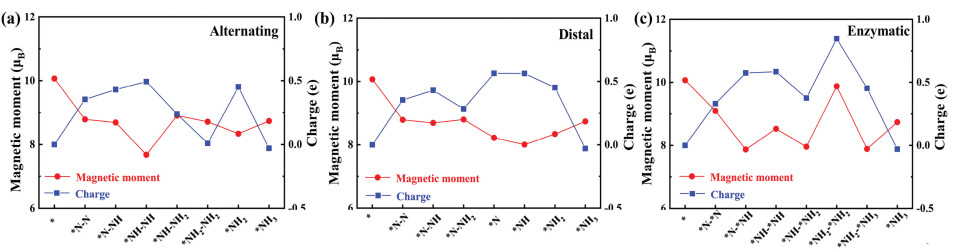
To explore the binding nature of the NRR mechanism, we have performed Bader charge analyses [56]. As shown in Figs. 5a-c, all the reaction species are negatively charged due to the high electronegativity of the N atom. The amounts of charge transfer are 0.354e and 0.331e for N2 adsorbed with end-on and side-on configurations, respectively. We also plotted the charge difference density diagram, which can be used to analyse the bonding properties and the orbital properties of the electron cloud. The charge accumulation (yellow) is distributed around two N atoms and Fe-N bonds, while the charge depletion (cyan) is distributed around the N-N bonds and the bonded surface metal atoms could be observed in two adsorption configurations (Fig. S3 in Supporting information). This conclusion supports our calculation results. The charge transfer revealed that 0.331e is injected into *N2, which effectively activated *N2. In addition, due to the formation of the Fe-N bond, the separated electrons in the Fe atoms are donated into the anti-bonding orbital, forming an electron transfer route that weakened the N≡N bond and made the subsequent hydrogenation easier. Generally speaking, more charge transfer corresponds to larger adsorption energy. Although our charge transfer and adsorption energy results do not completely conform to a linear relationship, the results do support a weak positive correlation between charge transfer and adsorption energy. Based on the information in Table 1 and Fig. 5, it can be concluded that strong adsorption indicated larger adsorption energy and more charge transfer. The results are as follows: −2.390 eV and 0.493e of *NH-NH@Fe2B2 for the alternating mechanism; −4.229 eV and 0.564e of *N@Fe2B2 for the distal mechanism; −2.522 eV and 0.848e of *NH2-*NH2@Fe2B2 for the enzymatic mechanism. Previous studies have reported that the donated electrons reduced the strength of the N≡N triple bond and the energy barrier of the PDS of hydrogenation [9, 10]; this supports our calculated results, indicating that there is more charge transfer using the enzymatic mechanism. For example, the *N2H gained more electrons via the enzymatic mechanism than with any of the other two mechanisms, and these extra donated electrons are further transmitted into the antibonding orbital of *N2H to strengthen the *N2H binding, leading to a lower free energy (0.02 eV). Thus, the substantial electron transfer should significantly improve the electronic transport properties, which can improve the catalytic activity.

|
Download:
|
| Fig. 5. The total magnetic moments (μB) and charge transfer (e) for the NRR on Fe2B2 through (a) alternating, (b) distal and (c) enzymatic mechanisms, respectively. | |
{kind=link}
Upon adsorption of all the reaction species, the total magnetic moment is significantly reduced by 1–2 μB, the decline is more than 2 μB for some species. The magnetic moment is mostly influenced by the d orbitals of Fe atoms, which are not filled with electrons. Therefore, the main reason for the reduction of total magnetic moment is that the empty d orbitals in the Fe atoms could receive the lone-pair electrons of N2, which led to a decrease in the number of unpaired electrons in Fe atoms after adsorption. Finally, we compared the relationship between charge transfer and magnetic moment. Generally, greater charge transfer between atoms corresponds to more distinct orbital hybridisation, implying that the charge transfer and orbital hybridisation affect the magnetic moment of the entire system. In our calculation, considering the end-on configuration using either the alternating or distal mechanism, the charge transfer is indirectly proportional to the magnetic moment, which means that the two curves in Figs. 5a and b display opposing trends. In contrast to the end-on configuration, more charge transfer do not result in significant reduction in magnetic moment for the side-on configuration of the enzymatic mechanism. This can be attributed to the different adsorption site and the relatively lower limiting potential.
In summary, we have identified that the first hydrogenation step for generating *N2H is key, and we have elucidated the PDS for the entire NRR process via the alternating and distal mechanisms. However, these mechanisms possess very large ΔG values (*N–N + H+ + e− = *N–NH) (1.26 eV); hence, it is difficult to bind *N2H. In order to solve this problem, we attempted to change the catalyst, by introducing defects or doping heteroatoms. Effective use of vacancies is considered to be one of the most effective defect strategies to improve catalytic behaviours in the NRR [57]. It has been revealed that the presence of various vacancies can effectively regulate the intrinsic properties including the electronic structure, charge transport, and surface adsorption capacity [58, 59]. Consequently, we can remove the B atom nearest to the TFe atom on the surface of Fe2B2 to form a B vacancy and calculate the free energy for generating *N2H (*N-N + H+ + e- = *N-NH). ΔG =1.01 eV indicates that the free energy barrier is reduced by 0.25 eV due to the B-vacancy (Fig. S4a in Supporting information). In addition, doping with a heteroatom is considered another effective approach to tailor electronic structures, reduce the reaction barrier, and modify chemical composition of catalysts [60-62]. We chose Al, Ga, and In to act as heteroatoms; they all belong to the same main group as B. It is well known that the atomic number, the number of electron layers, the atomic radius, the ease of electron loss and the metallicity of elements for the same group increase gradually from top to bottom. Increasing ease of electron loss played a positive role in weakening the N≡N triple bond, and this can help to reduce the ΔG by 0.26 eV, 0.06 eV and 0.10 eV after doping with Al, Ga and In, respectively (Figs. S4b-d in Supporting information).
Furthermore, the change in N-N bond length could also indicate the activation degree of N2. The N-N bond length of N2 and N2H adsorbed on pristine, defective, and doped Fe2B2 are summarised in Table S1 (Supporting information). These data indicated that the N-N bonds are all stretched due to the introduction of vacancies and dopants, illustrating that N-N bonds are activated more effectively. Especially in the case of Al doping, the N-N bond length is elongated to 1.254 Å, consistent with the lower free energy barrier of 1.00 eV (Fig. S4b). Accordingly, these vacancies and dopants help improve the catalytic activity, which opened up new opportunities for us to design and develop highly efficient and highly selective catalysts for NRR under ambient conditions. In future, we will study and report the effects of vacancies and dopants in detail.
In conclusion, we have performed first-principles calculations to systematically investigate the catalytic performance of Fe2B2. The following results demonstrate that the enzymatic mechanism is more favourable: (1) The limiting potential of −0.44 V; (2) the lower free energy (only 0.02 eV) of the first hydrogenation step (*N–N + H+ + e− = *N–*NH); and (3) the increased electron transfer for the whole NRR process. Therefore, efficient and stable electrocatalysts of Fe2B2 can promote N2 adsorption and accelerate electron transfer using an enzymatic mechanism to activate the NRR with a low free energy. In addition, considering all the reaction species adsorption, the total magnetic moment is significantly reduced by 1–2 μB, the decline range of some species is more than 2 μB. Both vacancies and dopants can be helpful to reduce the reaction energy barrier of the potential-determining step. We also hope that the insights obtained from this theoretical study offer guidance for the design of new and efficient electrocatalysts for the NRR.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors thank the National Natural Science Foundation of China for financial support (Nos. 21603109, 11904081 and 21876104). This research is also supported by Henan Joint Funds of the National Natural Science Foundation of China (No. U1404216), the Special Fund of Tianshui Normal University, China (No. CXJ2020-08), the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0676).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.02.043.
| [1] |
H.P. Jia, E.A. Quadrelli, Chem. Soc. Rev. 43 (2014) 547-564. DOI:10.1039/C3CS60206K |
| [2] |
V. Smil, Nature 400 (1999) 415. DOI:10.1038/22672 |
| [3] |
M. Kitano, Y. Inoue, Y. Yamazaki, et al., Nat. Chem. 4 (2012) 934-940. DOI:10.1038/nchem.1476 |
| [4] |
M.A. van Kessel, D.R. Speth, M. Albertsen, et al., Nature 528 (2015) 555-559. DOI:10.1038/nature16459 |
| [5] |
M. Kitano, S. Kanbara, Y. Inoue, et al., Nat. Commun. 6 (2015) 6731. DOI:10.1038/ncomms7731 |
| [6] |
M.E. Volopin, V.B. Shur, E.G. Berkovich, Inorg. Chim. Acta Rev. 280 (1998) 264-274. DOI:10.1016/S0020-1693(98)00206-0 |
| [7] |
G.J. Leigh, Science 279 (1998) 506-507. DOI:10.1126/science.279.5350.506 |
| [8] |
J. Wang, Y.P. Liu, H. Zhang, et al., Catal. Sci. Technol. 9 (2019) 4248-4254. DOI:10.1039/C9CY00907H |
| [9] |
S. Wang, B. Li, L. Li, et al., Nanoscale 12 (2020) 538-547. DOI:10.1039/C9NR09157B |
| [10] |
C.Y. Ling, X.H. Niu, Q. Li, et al., J. Am. Chem. Soc. 140 (2018) 14161-14168. DOI:10.1021/jacs.8b07472 |
| [11] |
G.Z. Gu, K.Y. Wang, N.N. Xiong, et al., Dalton Trans. 48 (2019) 5083-5089. DOI:10.1039/C9DT00013E |
| [12] |
N. Zhang, L.Y. Hong, A.F. Geng, et al., Chin. Chem. Lett. 29 (2018) 1409-1412. DOI:10.1016/j.cclet.2017.12.003 |
| [13] |
D.D. Wang, Y.Q. Zou, L. Tao, et al., Chin. Chem. Lett. 30 (2019) 826-838. DOI:10.1016/j.cclet.2019.03.051 |
| [14] |
J. Chen, J. Zhan, Y.M. Zhang, et al., Chin. Chem. Lett. 30 (2019) 735-738. DOI:10.1016/j.cclet.2018.08.020 |
| [15] |
C.Z. He, R. Wang, H.Y. Yang, et al., Appl. Surf. Sci. 507 (2020) 145076. DOI:10.1016/j.apsusc.2019.145076 |
| [16] |
C.Z. He, R. Wang, D. Xiang, et al., Appl. Surf. Sci. 509 (2020) 145392. DOI:10.1016/j.apsusc.2020.145392 |
| [17] |
S. Zhang, Y. Zhao, R. Shi, et al., EnergyChem 1 (2019) 100013. DOI:10.1016/j.enchem.2019.100013 |
| [18] |
X.Y. Cui, C. Tang, Q. Zhang, Adv. Energy Mater. 8 (2018) 1800369. DOI:10.1002/aenm.201800369 |
| [19] |
L. Shi, Y. Yin, S.B. Wang, et al., ACS Catal. 10 (2020) 6870-6899. DOI:10.1021/acscatal.0c01081 |
| [20] |
P. Wang, F. Chang, W. Gao, et al., Nat. Chem. 9 (2017) 64-70. DOI:10.1038/nchem.2595 |
| [21] |
N. Cao, G. Zheng, Nano Res. 11 (2018) 2992-3008. DOI:10.1007/s12274-018-1987-y |
| [22] |
D. Bao, Q. Zhang, F.L. Meng, et al., Adv. Mater. 29 (2017) 1604799. DOI:10.1002/adma.201604799 |
| [23] |
W.Z. Fu, Y.D. Cao, Q.Y. Feng, et al., Nanoscale 11 (2019) 1379-1385. DOI:10.1039/C8NR08724E |
| [24] |
Z.W. Chen, J.M. Yan, Q. Jiang, Small Methods 180029 (2018) 1-8. |
| [25] |
X.H. Zhao, X. Lan, D.K. Yu, et al., Chem. Commun. 54 (2018) 13010-13013. DOI:10.1039/C8CC08045C |
| [26] |
S. Ji, Z.X. Wang, J.X. Zhao, J. Mater. Chem. A 7 (2019) 2392-2399. DOI:10.1039/C8TA10497B |
| [27] |
C.W. Liu, Q.Y. Li, J. Zhang, et al., J. Mater. Chem. A 7 (2019) 4771-4776. DOI:10.1039/C8TA08219G |
| [28] |
J. Xie, H.L. Dong, X.H. Cao, et al., Mater. Chem. Phys. 243 (2020) 122622. DOI:10.1016/j.matchemphys.2020.122622 |
| [29] |
X.Y. Guo, S.P. Huang, Electrochim. Acta 284 (2018) 392-399. DOI:10.1016/j.electacta.2018.07.168 |
| [30] |
Y.M. Qian, Y.Y. Liu, Y. Zhao, et al., EcoMat 2 (2020) e12014. |
| [31] |
Y. Yang, J. Liu, Z. Wei, et al., ChemCatChem 11 (2019) 2821-2827. DOI:10.1002/cctc.201900536 |
| [32] |
H. Shi, M. Xia, L.T. Jia, et al., Chem. Phys. 536 (2020) 110783. DOI:10.1016/j.chemphys.2020.110783 |
| [33] |
X.R. Zhao, F.X. Yin, N. Liu, et al., J. Mater. Sci. 52 (2017) 10175-10185. DOI:10.1007/s10853-017-1176-5 |
| [34] |
S.M. Chen, S. Perathoner, C. Ampelli, et al., Angew. Chem. Int. Ed. 56 (2017) 2699-2703. DOI:10.1002/anie.201609533 |
| [35] |
H. Du, C.Z. Yang, W.H. Pu, et al., ACS Sustainable Chem. Eng. 8 (2020) 10572-10580. DOI:10.1021/acssuschemeng.0c03675 |
| [36] |
L. Fu, R. Wang, C.X. Zhao, et al., Chem. Eng. J. 414 (2021) 128857. DOI:10.1016/j.cej.2021.128857 |
| [37] |
Q.L. Liu, S.N. Wang, G.L. Chen, et al., Inorg. Chem. 58 (2019) 11843-11849. DOI:10.1021/acs.inorgchem.9b02280 |
| [38] |
M.A. Legare, G. Belanger-Chabot, R.D. Dewhurst, et al., Science 359 (2018) 896-899. DOI:10.1126/science.aaq1684 |
| [39] |
W. Qiu, X.Y. Xie, J. Qiu, et al., Nat. Commun. 9 (2018) 3485. DOI:10.1038/s41467-018-05758-5 |
| [40] |
Q.Y. Li, C.W. Liu, S.Y. Qiu, et al., J. Mater. Chem. A 7 (2019) 21507-21513. DOI:10.1039/C9TA04650J |
| [41] |
J.R. Huo, L. Fu, C.X. Zhao, et al., Chin. Chem. Lett. 32 (2021) 2269-2273. DOI:10.1016/j.cclet.2020.12.059 |
| [42] |
D.W. Zhou, C.P. Li, F.R. Yin, et al., Chin. Chem. Lett. 31 (2020) 2325-2329. DOI:10.1016/j.cclet.2020.04.045 |
| [43] |
C.Y. Pu, J.H. Yu, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 1081-1085. DOI:10.1016/j.cclet.2020.08.042 |
| [44] |
Y. Song, X.Y. Li, C.Z. He, Chin. Chem. Lett. 32 (2021) 1106-1110. DOI:10.1016/j.cclet.2020.08.024 |
| [45] |
H.L. Hu, H. Yang, X.Y. Yang, et al., Chin. Chem. Lett. 31 (2020) 3213-3215. DOI:10.1016/j.cclet.2020.08.025 |
| [46] |
X. Fu, H.Y. Yang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 1089-1094. DOI:10.1016/j.cclet.2020.08.031 |
| [47] |
L. Chen, C.Z. He, R. Wang, et al., Chin. Chem. Lett. 32 (2021) 53-56. DOI:10.1016/j.cclet.2020.11.013 |
| [48] |
G. Kresse, J. Hafner, Phys. Rev. B 47 (1993) 558-561. DOI:10.1103/PhysRevB.47.558 |
| [49] |
G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [50] |
P.E. Blochl, Phys. Rev. B 50 (1994) 17953-17979. DOI:10.1103/PhysRevB.50.17953 |
| [51] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [52] |
J.K. Norskov, J. Rossmeisl, A. Logadottir, et al., J. Phys. Chem. B 108 (2004) 17886-17892. DOI:10.1021/jp047349j |
| [53] |
L. Zhang, X.Q. Ji, X. Ren, et al., Adv. Mater. 30 (2018) 1800191. DOI:10.1002/adma.201800191 |
| [54] |
L. Zhang, X.Q. Ji, X. Ren, et al., ACS Sustain. Chem. Eng. 6 (2018) 9550-9554. DOI:10.1021/acssuschemeng.8b01438 |
| [55] |
X. Ren, G.W. Cui, L. Chen, et al., Chem. Commun. 54 (2018) 8474-8477. DOI:10.1039/C8CC03627F |
| [56] |
D.W. Ma, T.X. Li, Q.G. Wang, et al., Carbon 95 (2015) 756. DOI:10.1016/j.carbon.2015.09.008 |
| [57] |
C.L. Mao, J.X. Wang, Y.J. Zou, et al., Green Chem. 21 (2019) 2852-2867. DOI:10.1039/C9GC01010F |
| [58] |
K. Chu, Y.P. Liu, Y.H. Cheng, et al., J. Mater. Chem. A 8 (2020) 5200-5208. DOI:10.1039/D0TA00220H |
| [59] |
C. Lv, Y.M. Qian, C.S. Yan, et al., Angew. Chem. Int. Ed. 57 (2018) 10246-10250. DOI:10.1002/anie.201806386 |
| [60] |
C.N.R. Rao, K. Gopalakrishnan, A. Govindaraj, Nano Today 9 (2014) 324-343. DOI:10.1016/j.nantod.2014.04.010 |
| [61] |
K. Chu, Y.P. Liu, Y.B. Li, et al., Appl. Catal. B: Environ. 264 (2020) 118525. DOI:10.1016/j.apcatb.2019.118525 |
| [62] |
N. Zhang, A. Jalil, D.X. Wu, et al., J. Am. Chem. Soc. 140 (2018) 9434-9443. DOI:10.1021/jacs.8b02076 |