 2021, Vol. 32
2021, Vol. 32 

b State Key Laboratory of Biotherapy, West China Hospital, West China Medical School, Sichuan University, Chengdu 610041, China
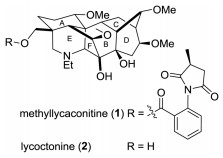
C19-Diterpenoid alkaloids, mainly isolated from the genera Aconitum and Delphinium, constitute a structurally diverse class of natural products and have been shown a wide range of potent physiological activities [1]. For example, methyllcaconitine (MLA) (1) (Fig. 1), first discovered from Delphinium brownii by Manske in 1938 [2] and its whole stereochemical structure was correctly determined by Pelletier in 1981 [3], is a representative C19-diterpenoid alkaloid and major toxic component found in at least 30 Delphinium species as well as in Consolida ambigua and Inaularoyaleana [4]. The toxicity has been attributed to its ability to act at the neuromuscular junction, inhibiting neurotransmission and inducing paralysis [5], and it has been found to be a potent inhibitor of the nicotinic acetylcholine receptor (nAChR) binding in mammalian and insect neural membranes [5]. Among the α-7 subtype nAChR, MLA is one of the most effective small molecule competitive antagonists reported so far. Researchers have proposed that MLA or analogs could be potentially useful for developing drug candidates for the treatment of neurological conditions such as schizophrenia, Alzheimer's disease and epilepsy [6].

|
Download:
|
| Fig. 1. Methyllycaconitine and lycoctonine. | |
{kind=link}
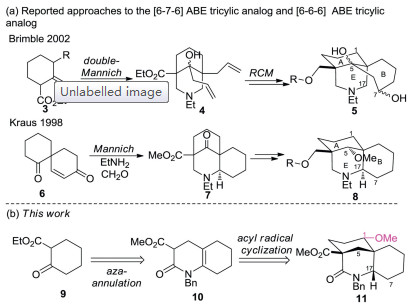
Structure activity relationship (SAR) studies on MLA (1) have shown that the N-substituted anthranilate ester moiety and a homocholine backbone corresponding to the piperidine E ring of lycoctonine (2) are essential for potent pharmacological activities [7]. Several research groups [8], including our own [9], have synthesized a variety of analogues of MLA including E, AE, BE, ABE, AEF, ABDE, ABEF and ABDEF rings, some of which display significant biological activities. Of particular interest to our group is the [6-6-6] ABE tricyclic ring system of MLA due to the resembled [6-7-6] ABE tricyclic analogue 5 (one of diastereomers) [10] has been reported to exhibit high nAChR affinity binding (IC50 = 478 nmol/L) which is amongst the highest reported to date for small molecule analogues of MLA. Although Kraus and co-workers have reported the preparation of [6-6-6] ABE tricyclic analogue 8 [11] from a known spirocyclic diketone 6, no structure-activity data was given and compounds with a key oxygenated substitution at the C-1 position had not been included (Scheme 1). It has been found that substitution at the C-1has a considerable effect on biological activity, so that compounds with a C-1hydroxyl group, e.g., grandiflorine, exhibits 100–1000 times less binding efficiency for nAChR than MLA with a C-1 methoxy group [7b]. In order to explore new area of structural alteration of MLA with the aim to obtain more potent nAChRs antagonists, herein, we described a new route for the synthesis of the [6-6-6] ABE tricyclic analogue 11 bearing a key methoxy group at the position corresponding to C-1 of MLA, utilizing an efficient aza-annulation and a transannular acyl radical cyclization as the key steps (Scheme 1).

|
Download:
|
| Scheme 1. (a) Representative approaches to the ABE analogues of methylly-caconitine; (b) an aza-annulation followed by acyl radical cyclization strategy in this work. | |
{kind=link}
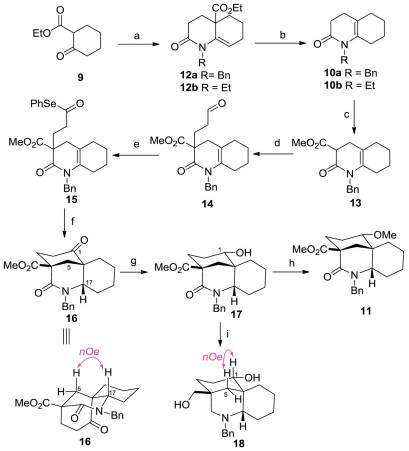
The synthetic study commenced with the preparation of the known N-alkyl substituted 3, 4, 5, 6, 7, 8-hexahydro-2(1H)-quinolinone 10 [12, 13] as the precursor of BE rings (Scheme 2). Although a principle method for preparation of this crucial type of ene-lacatams has been reported starting from cyclohexone and acrylonitrile by Murahashi via a two-step sequence [13], poor yield and using of noble metal catalyst (RuH2PPh3) under forcing conditions (sealed tube, 120 ℃) limit its application in a scalable synthesis. Other methodology [12] involving aza-annulation with imines and various acrylate derivatives were restricted by lack of alkene regioselectivity and poor yields that result from formation of the imines from ketones. In this event, we present a new two-step reaction to produce 10 involving a highly efficient aza-annulation of a β-enamino ketone and an incidentally discovered enamine tautomerization induced by a facile decarboxylation. First, following the Stille's aza-annulation protocol [14], reaction of ethyl 2-cyclohexanonecarboxylate 9 with benzyl amine in the presence of the Et2O·BF3 followed by exposure of resultant crude enamine to acryloyl chloride gave the 12a smoothly, with the alkene exocyclic relative to the ring generated through annulation. Then hydrolysis of the ester group of 12a provided the crude acid, which without purification, was warmed at 50 ℃ under acidic conditions in one pot to effect a mild decarboxylation, delivering the desired double bond shifting ene-lacatam10a in 81% yield over two steps. Pleasingly, replacement of benzyl amine to ethyl amine in first step could achieve the almost the same yield of N-ethyl substituted ene-lacatam 10b by executing the same two-step reaction sequence.

|
Download:
|
| Scheme 2. Synthesis of ABE ring analogues 18 and 19. Reagents and conditions: (a) benzylamine or ethylamine, Et2O·BF3, CHCl3, reflux, 12 h; then acrylylchloride, THF, reflux, 12 h, 95% for 12a, 94% for 12b; (b) KOH, EtOH/H2O (3:1, v/v), 50 ℃, 15 h; then 1 mol/L HCl, 50 ℃, 2 h, 85% for 10a; 84% for 10b; (c) LiHMDS, CH3OOCCl, THF, -78 ℃, 3 h, 80%; (d) acrolein, K2CO3, acetone, r.t., 12 h, 90%; (e) (ⅰ) 2-methyl-2-butene, NaClO2, NaH2PO4, t-BuOH/H2O (3:1, v/v), 50 ℃, 3 h; (ⅱ) Ph2Se2, n-Bu3P, DCM, 0 ℃, 1 h, 80% for two steps; (f) Bu3SnH, ACN, toluene, reflux, 3 h, 84%; (g) t-BuNH2·BH3, DCM, r.t., 2 h, 86%; (h) Ag2O, CH3I, sealed, 60 ℃, 24 h, 90%; (i) BH3·THF, THF, reflux, 2 h, 85%. | |
{kind=link}
With the BE-ring surrogate in place, the formation of bridged A-ring moiety with oxygen functionality at C-1 was addressed next by means of a pivotal intramolecular transannular cyclization via an acyl radical strategy. Thus, treatment of 10a with ClCOOMe in the presence of LiHMDS at -78 ℃ provided the β-ketoester 13 in 80% yield. Michael addition of 13 to acrolein gave the adduct 14 in 90% yield. Pinnick oxidation [15] cleanly converted aldehyde into crude acid, which without purification, was transformed into selenoester 15 through a selenation [16] using a combination of diphenyldiselenide and tributylphosphine in 80% yield over two steps. Having selenoester 15 in hand, we examined the key transannular radical cyclization [17] to construct the bridged analogues of ABE rings. Treatment of compound 15 with tributyltin hydride (1.5 equiv.) and 1, 10-azobiscyclohexanecarbonitrile (ACN) (0.2 equiv.) in toluene at reflux caused a transannular cyclization of an intermediary acyl radical to give the desired azabicyclo [3.3.1] compound 16 in a completely stereoselective manner. The stereochemistry of the radical addition could be confirmed with NOE difference experiments showing through-space interactions between the amino methine proton (H-17) and the adjacent methylene (H-5a) (the NOE spectrum of 16, see Supporting information). The exclusive formation of the trans-fused BE moiety in the present cyclization might be due to the acyl radical cyclization has ostensibly tended to be thermodynamically controlled [17d].
Next, incorporation of the functionality on the ABE rings was attempted. To our surprising, selective reduction of the ketone group of 16 was unexpectedly difficult. Employment of standard reductive reagents such as NaBH4, NaBH(OAc)3 or L-selectride uniformly gave a mixture of unidentified products. After screening a variety of other conditions, it was finally discovered that treatment of ketone with tert-butylamine-borane complex [18] at room temperature afforded the desired alcohol 17 as a single stereo isomer. Methylation with methyl iodide in the presence of silver(I) oxide [19] as soft base in THF at reflux proceeded cleanly to furnish 11 in 90% yield. Subsequent thorough hydride reduction of the ester and lactam moieties of 17 with BH3 in THF [20] provided diol 18 in 85% yield. The stereochemistry of the OH group at C-1 of 17 was established as an α orientation on the basis of the observation of the key NOESY relationship between H-1 and H-5β in 18 (the NOE spectrum of 18, see Supporting information).
In summary, the synthesis of the new [6-6-6] ABE tricyclic ring analogues of MLA with C-1 oxygenated substituents has been achieved. The improved two-step reaction sequence involving a smooth aza-annulation of β-enamino ketone and the facile decarboxylation under mild condition provided a highly efficient and practical preparation of N-alkyl substituted ene-lactam as the precursor of BE rings. The bridged A ring unit was constructed through a convenient transannular acyl radical cyclization with concomitant equipment of the key C-1 oxygen functionality. Elaboration of the C-1 methoxy group was achieved by selective reduction followed by methylation. Further preparation of ABE tricyclic ring analogues with appendage of a methylsuccinimidoanthranilate moiety or other N-alkyl derivatives, and the evaluation of these compounds and intermediates at the α7nAChR subtype for binding affinity, are currently being explored and will be reported in due course.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge National Natural Science Foundation of China (Nos. 21472129 and 21871190) for financial support of this work by grants.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.03.068.
| [1] |
(a) F.P. Wang, Q.H. Chen, The C19-diterpenoid alkaloids, in: G.A. Cordell (Ed.), The Alkaloids: Chemistry and Biology, Academic Press Inc, San Diego, 2010, pp. 1-577.
|
| [2] |
R.H.F. Manske, Can. J. Res. 16B (1938) 57-63. DOI:10.1139/cjr38b-007 |
| [3] |
S.W. Pelletier, N.V. Mody, K.I. Varughese, J.A. Maddry, H.K. Desai, J. Am. Chem. Soc. 103 (1981) 6536-6538. DOI:10.1021/ja00411a062 |
| [4] |
(a) S.W. Pelletier, N.V. Mody, B.S. Joshi, L.C. Schramm, l3C and proton NMR shift assignments and physical constants of C19-diterpenoid alkaloids, in: S.W. Pelletier (Ed. ), Alkaloids: Chemical and Biological Perspectives, SpringerVerlag New York Inc., New York, 1984, pp. 205-213; (b) S.W. Pelletier, B.S. Joshi, Carbon-13 and proton NMR shift assignments and physical constants of norditerpenoid alkaloids, in: S.W. Pelletier (Ed. ), Alkaloids: Chemical and Biological Perspectives, Springer-Verlag New York Inc., New York, 1991, pp. 297-301. |
| [5] |
S. Wonnacott, E.X. Albuquerque, D. Bertrand, Methods Neurosci. 12 (1993) 263-267. |
| [6] |
(a) J. Malysz, J.H. Gronlien, D.J. Anderson, et al., J. Pharmacol. Exp. Ther. 330 (2009) 257-263; (b) S.N. Haydar, J. Dunlop, Curr. Top. Med. Chem. 10 (2010) 144-147. |
| [7] |
(a) K.R. Jennings, D.G. Brown, D.P. Wright, Experientia 42 (1986) 611-613; (b) C.F. Kukel, K.R. Jennings, Can. J. Physiol. Pharmacol. 72 (1994) 104-107; (c) D.R.E. Macallan, G.G. Lunt, S. Wonnacott, et al., FEBS Lett. 226 (1988) 357-363; (d) M. Reina, A. Gonzalez-Coloma, Phytochem. Rev. 6 (2007) 81-95. |
| [8] |
(a) K.J. Goodall, D. Barker, M.A. Brimble, Synlett (2005) 1809-1813; (b) J. Huang, C.M. Orac, S. McKay, D.B. McKay, S.C. Bergmeier, Bioorg. Med. Chem. 16 (2008) 3816-3820; (c) Y. Chan, J. Balle, S.J. Kevin, et al., Tetrahedron 66 (2010) 7179-7184; (d) H. Guthmann, D. Conole, E. Wright, et al., Eur. J. Org. Chem. (2009) 1944-1947; (e) J. Huang, S.C. Bergmeier, Tetrahedron 64 (2008) 6434-6437; (f) A. Lehmann, C. Brocke, D. Barker, M.A. Brimble, Eur. J. Org. Chem. (2006) 3205-3210; (g) E. Dickson, L.I. Pilkington, M.A. Brimble, Tetrahedron 72 (2016) 400-414; (h) K.J. Sparrow, S. Carley, T. Sohnel, D. Barker, M.A. Brimble, Tetrahderon 71 (2015) 2210-2221; (i) K.J. Goodall, M.A. Brimble, D. Barker, Tetrahedron 68 (2012) 5759-5778; (j) K.J. Sparrow, D. Barker, M.A. Brimble, Tetrahedron 68 (2012) 1017-1028; (k) K.J. Sparrow, D. Barker, M.A. Brimble, Tetrahedron 67 (2011) 7989-7999. |
| [9] |
(a) Z.G. Liu, H. Cheng, M.J. Ge, L. Xu, F.P. Wang, Tetrahedron 69 (2013) 5431-5435; (b) H. Cheng, F.H. Zeng, D. Ma, et al., Org. Lett. 16 (2014) 2299-2301; (c) M.C. Liu, C.X. Cheng, W.Y. Xiong, et al., Org. Chem. Front. 5 (2018) 1502-1505. |
| [10] |
A.R.L. Davies, D.J. Hardick, I.S. Blagbrough, et al., Biochem. Soc. Trans. 25 (1997) 545-548. DOI:10.1042/bst025545s |
| [11] |
A.K. George, D. Elena, Tetrahedron Lett. 39 (1998) 2451-2454. DOI:10.1016/S0040-4039(98)00239-1 |
| [12] |
(a) I. Ninomiya, T. Naito, S. Higuchi, J. Chem. Soc. Chem. Comm. 24 (1970) 1662-1662; (b) R. Shabana, J.B. Rasmussen, S.O. Olesen, S.O. Lawesson, Tetrahedron 36 (1980) 3047-3051; (c) A.A. El-Barbary, S. Carlsson, S.O. Lawesson, Tetrahedron 38 (1982) 405-412; (d) L. Stanislaw, P. Beata, Syn. Commun. 32 (2002) 875-880; (e) W.F. Dai, C.H. Wang, X. Zhang, J.M. Zhang, M.D. Lang, Sci. China Chem. 51 (2008) 1044-1050. |
| [13] |
(a) M. Shunichi, S. Shigehiro, S. Eiichiro, N. Takeshi, J. Org. Chem. 57 (1992) 2521-2523; (b) M. Shunichi, S. Shigehiro, S. Eiichiro, N. Takeshi, Tetrahedron 49 (1993) 8805-8826. |
| [14] |
(a) K. Paulvannan, J.R. Stille, J. Org. Chem. 59 (1994) 1613-1620; (b) N.S. Barta, A. Brode, J.R. Stille, J. Am. Chem. Soc. 116 (1994) 6201-6206. |
| [15] |
B.S. Bal, J.W.E. Childers, H.W. Pinnick, Tetrahedron 37 (1981) 2091-2096. DOI:10.1016/S0040-4020(01)97963-3 |
| [16] |
S. Masamune, Y. Hayase, W. Schilling, W.K. Chan, G.S. Bates, J. Am. Chem. Soc. 99 (1977) 6756-6758. DOI:10.1021/ja00462a049 |
| [17] |
(a) C. Chatgilialoglu, D. Crich, M. Komatsu, I. Ryu, Chem. Rev. 99 (1999) 1991-2070; (b) C.H. Schiesser, U. Wille, H. Matsubara, I. Ryu, Acc. Chem. Res. 40 (2007) 303-313; (c) K. Yoshikai, T. Hayama, K. Nishimura, K. Yamada, K. Tomioka, J. Org. Chem. 70 (2005) 681-683; (d) W.S. Grant, K. Zhu, S.L. Castle, Org. Lett. 8 (2006) 1867-1870; (e) M. Inoue, Y. Ishihara, S. Yamashita, M. Hirama, Org. Lett. 8 (2006) 5801-5804; (f) T. Roca, M.L. Bennasar, J. Org. Chem. 76 (2011) 4213-4218; (g) H. Zaimoku, T. Taniguchi, H. Ishibashi, Org. Lett. 14 (2012) 1656-1658. |
| [18] |
I. Takashi, T. Tamaaki, C.C. Frederic, G. Junichi, N. Toshio, J. Lipid Res. 32 (1991) 649-658. DOI:10.1016/S0022-2275(20)42052-8 |
| [19] |
L.C. Baillie, J.R. Bearder, W.S. Li, J.A. Sherringham, D.A. Whiting, J. Chem. Soc. Perkin Trans. 1 (1998) 4047-4050. |
| [20] |
(a) R.C. Neil, H.D. Suzanne, G. Matthew, et al., Org. Process Res. Dev. 19 (2015) 865-871; (b) H. Gregory, K. Masanari, L.B. Stephen, J. Am. Chem. Soc. 125 (2003) 11253-11258. |