 2021, Vol. 32
2021, Vol. 32 


b Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
Organofluorine compounds are of great importance in the fields of medicine, agricultural chemistry, and functional materials [1, 2]. The trifluoromethylthio (–SCF3) group is of particular interest because its highly tunable lipophilicity, metabolic stability, and strongly electron-withdrawing nature (Hansch parameter πR = 1.44) [3] make it useful for fine-tuning the pharmacological and pharmacokinetic properties of drug candidates [4, 5]. Consequently, reagents and methods for efficiently incorporating trifluoromethylthio groups into target molecules have attracted considerable interest in recent years [6]. However, the number of methods for synthesis of trifluoromethylthioesters is rather limited. In pioneering work, the groups of Man [7a], Yagupolskii [7b], and Chen and Weng [7c] showed that trifluoromethylthioesters can be synthesized by reactions of acid chlorides with Hg(SCF3)2, NMe4SCF3, or (bpy)CuSCF3 (Scheme 1A). Because Hg(SCF3)2 is highly toxic, NMe4(SCF3) is relatively unstable, and stoichiometric amounts of metallic reagents are required, the scope and utility of these reactions are limited.

|
Download:
|
| Scheme 1. Trifluoromethylthiolation reactions. | |
We speculated that trifluoromethylthioesters could be synthesized by a method involving the use of photoredox catalysis which has recently attracted much interest among synthetic chemists [8]. The Glorius group [9] reported an elegant method for obtaining trifluoromethylthioesters from aldehydes via a hydrogen atom transfer (HAT) process (Scheme 1B), but a noble metal photoredox catalyst and a HAT mediator are required. The direct use of an inexpensive HAT photo catalyst would be a more reagent- and redox-economical strategy. More recently, the Wang group [10] reported a method for visible-light-promoted synthesis of trifluoromethylthioesters from aldehydes, but heat and a stoichiometric amount of a radical initiator are needed to trigger the radical process, making the reaction relatively harsh (Scheme 1B). Because aldehydes are economical and readily available, we explored their trifluoromethylthiolation under mild conditions using inexpensive and readily available metal photocatalysts (Scheme 1C).
We envisioned that polyoxometalates (many of which have high-energy excited states and can accomplish the required C-H abstraction) [11] would be ideal HAT catalysts. We were particularly interested in tetrabutylammonium decatungstate (TBADT, (nBu4N)4[W10O32]), a highly efficient HAT photocatalyst that is widely used for dehydrogenation, oxidation, and conjugate addition reactions of unactivated aliphatic C-H bonds [12]. We recently developed a protocol for photoredox deuteration and difluoromethylthiolation reactions of aldehydes mediated by TBADT, which is responsible for the generation of an acyl radical [13]. Herein we report that we have succeeded in developing a protocol that uses TBADT as a HAT photocatalyst for efficient synthesis of trifluoromethylthioesters by direct functionalization reactions of aldehydic C(O)-H bonds.
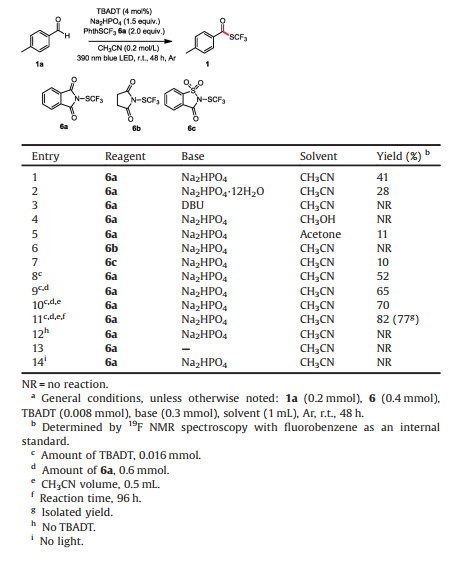
The proposed mechanism for the reaction is shown in Scheme 2. The reaction is initiated by photoexcitation of TBADT (2) followed by intersystem crossing to produce triplet excited state 3, which has a lifetime of 55 ns [14] and serves as the HAT catalyst. Excited state 3 selectively abstracts the formyl H atom of 4-methylbenzaldehyde (1a) to afford singly reduced decatungstate 4 and nucleophilic formyl radical 5. Radical 5 then readily reacts with the electrophilic N-(trifluoromethylthio)phthalimide (PhthSCF3, 6a) to afford desired product 1 upon elimination of a phthalimide radical Phth• (7). Finally, with Na2HPO4 as a base, 7 is reduced by decatungstate 4 to 8, with simultaneously regeneration of the active HAT photocatalyst 2.

|
Download:
|
| Scheme 2. Proposed mechanism of the trifluoromethylthiolation reaction. | |
We began our study by using 1a and 6a as model substrates to optimize the reaction conditions (Table 1). We were pleased to find that in the presence of 4 mol% TBADT and 1.5 equiv. of disodium phosphate as a base, irradiation of an acetonitrile solution of 1a and 6a with a 390 nm blue LED at room temperature under argon provided a 41% yield of desired product 1 (entry 1). Screening of various alternative bases revealed that Na2HPO4·12H2O gave a low yield, and DBU (1, 8-diazabicyclo[5.4.0]undec-7-ene) failed to afford any of the target product (entries 2 and 3). Methanol and acetone proved to be inferior solvents to CH3CN (entries 4 and 5). Other SCF3 sources (6b and 6c) gave lower yields than 6a (entries 6 and 7). We also varied the catalyst loading, SCF3 reagent stoichiometry, solvent volume, and reaction time (entries 8–11) gratifyingly, these experiments greatly improved the yield (entry 11). Control experiments demonstrated that TBADT, base, and light were all essential for the success of the reaction (entries 12–14).
|
|
Table 1 Optimization of the reaction conditions. |
{kind=link}
{kind=link}
With satisfactory conditions (Table 1, entry 11) in hand, we explored the scope of the reaction with respect to the aldehyde (Scheme 3). First, we tested aldehyde substrates with various substituents on the aromatic ring and found that we could obtain the corresponding trifluoromethylthioesters. For example, para-substituted aryl aldehydes gave target products 1 and 9–20 in 24%–94% yields, and meta-substituted aldehydes gave 21–25 in yields of 35%–75%. In general, substrates with electron-donating groups gave better yields than those with electron-neutral groups. Notably, reaction of an aldehyde bearing a tertiary C-H bond selectively gave 10 (67% isolated yield), the product of C-H trifluoromethylthiolation at the aldehydic C(O)-H bond; no products of CO dissociation were observed [15]. Aryl aldehydes containing alkyl groups (1, 9–11), ethers (13–18), phenyl groups (12 and 22), and an ester (30) were found to be compatible with the reaction conditions, giving the corresponding products in moderate to high yields. Interestingly, several relatively sensitive yet versatile functional groups—boronic esters (19 and 25) and an alkene (20)—tolerated the trifluoromethylthiolation conditions well, which shows the potential utility of this protocol for medicinal and synthetic chemistry applications. Polysubstituted aldehydes were also suitable substrates (26–31), and naphthaldehydes afforded target products 32 and 33 in yields of 37% and 55%, respectively. Aware of the important role of heteroaromatic moieties in drug compound scaffolds, we were pleased to find that under our trifluoromethylthiolation conditions, N-heterocyclic substrates were also tolerated, giving 34 and 35 in 42% and 32% yield. However, our experiments uncovered a limitation of this reaction: substrates containing electron-withdrawing groups or ortho substituents on the phenyl ring were incompatible with the reaction conditions, possibly because of the steric bulk of TBADT and its tendency to react with nucleophilic sites.

|
Download:
|
| Scheme 3. Substrate scope with respect to the aldehyde. | |
{kind=link}
Because of the biological relevance of fluoroalkylthio groups [16, 4b], a mild, selective method for late-stage introduction of trifluoromethylthio groups into complex molecules is highly desirable for drug modification studies. We found that our protocol could readily be used for trifluoromethylthiolation of aromatic aldehydes such as fenbufen and ibuprofen derivatives to afford 36 and 37 (Scheme 4A). The successful late-stage functionalization of these two drug derivatives, which contain ester and carbonyl groups and a tertiary C-H bond, demonstrates the high chemoselectivity and functional group tolerance of our method.

|
Download:
|
| Scheme 4. Applications of the protocol. | |
{kind=link}
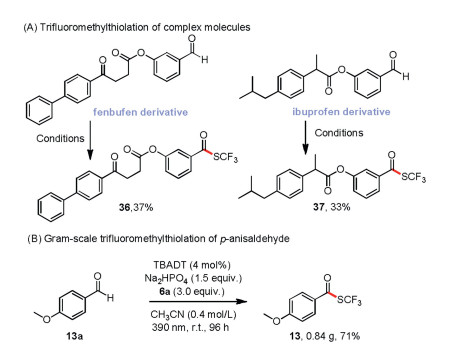
To demonstrate the viability of the method for preparative purposes, we carried out a gram-scale trifluoromethylthiolation reaction of p-anisaldehyde (5 mmol) under the standard conditions, except that the amount of catalyst was reduced to 4 mol%; the reaction was complete within 96 h and gave 13 in 71% yield (Scheme 4B).
Next we conducted some mechanistic studies (Scheme 5) in support of the proposed pathway shown in Scheme 2. When TEMPO ((tetramethylpiperidin-1-yl)oxyl) or 1, 1-diphenylethylene was used as a radical scavenger, the formation of 13 was completely inhibited. TEMPO adduct 2, 2, 6, 6-tetramethylpiperidin-1-yl 4-methoxybenzoate (38) was isolated in 21% yield, and radical trapping product 1-(4-methoxyphenyl)-3, 3-diphenylprop-2-en-1-one (39) was detected by mass spectrometry. These results suggest that acyl radical 5 was generated, and they clearly point to a radical pathway. Moreover, a light/dark experiment showed that product 13 did not form in the dark (Supporting information), which implies that light was essential and that any chain propagation process was short-lived.

|
Download:
|
| Scheme 5. Mechanistic experiments. | |
{kind=link}
In summary, we have developed a versatile photoredox-mediated HAT protocol for direct trifluoromethylthiolation of aldehydes. The mild reaction occurred at room temperature with the aldehyde as the limiting reagent, tolerated various functional groups, and had a broad substrate scope. Commercially available, stable aldehydes were used as the carbonyl source, and the HAT photocatalyst was TBADT, an inexpensive complex of an earth-abundant metal. This expedient alternative to traditional methods of synthesizing trifluoromethylthioesters can be expected to be useful for late-stage functionalization of natural products and drug molecules.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgmentsWe are grateful to the National Natural Science Foundation of China (Nos. 21732002, 22077071) for generous financial support for our programs.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.03.070.
| [1] |
(a) V.N. Boiko, Beilstein J. Org. Chem. 6 (2010) 880-921; (b) F. Toulgoat, S. Alazet, T. Billard, Eur. J. Org. Chem. (2014) 2415-2428; (c) X.H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115 (2015) 731-764; (d) C. Ni, J. Hu, Chem. Soc. Rev. 45 (2016) 5441-5454. |
| [2] |
(a) D.T. Wong, K.W. Perry, F.P. Bymaster, Nat. Rev. Drug Discov. 4 (2005) 764-774; (b) W.K. Hagmann, J. Med. Chem. 51 (2008) 4359-4369; (c) K.L. Kirk, Org. Process Res. Dev. 12 (2008) 305-321; (d) N.A. Meanwell, J. Med. Chem. 54 (2011) 2529-2591. |
| [3] |
(a) A. Leo, C. Hansch, D. Elkins, Chem. Rev. 71 (1971) 525-616; (b) L.M. Yagupol'skii, A.Y. Ilchenko, N.V. Kondratenko, Russ. Chem. Rev. 43 (1974) 32. |
| [4] |
(a) S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320-330; (b) J. Wang, M. Sànchez-Rosello', J.L. Aceña, etal., Chem. Rev. 114 (2014)2432-2506. |
| [5] |
C. Hansch, A. Leo, R.W. Taft, Chem. Rev. 91 (1991) 165-195. DOI:10.1021/cr00002a004 |
| [6] |
(a) T. Umemoto, S. Ishihara, J. Am. Chem. Soc. 115 (1993) 2156-2164; (b) I. Kieltsch, P. Eisenberger, A. Togni, Angew. Chem., Int. Ed. 46 (2007) 754-757; (c) F. Baert, J. Colomb, T. Billard, Angew. Chem., Int. Ed. 51 (2012) 10382-10385; (d) B. Langlois, D. Montégre, N. Roidot, J. Fluorine Chem. 68 (1994) 63-66; (e) C. KimmyCao, E. Tretyakov, C. Chen, Green Synth. Catal. 2 (2021) 62-65. |
| [7] |
(a) E.H. Man, D.D. Coffman, E.L. Muetterties, J. Am. Chem. Soc. 81 (1959) 3575-3577; (b) M.M. Kremlev, W. Tyrra, D. Naumann, Y.L. Yagupolskii, Tetrahedron Lett. 45 (2004) 6101-6104; (c) M. Zhang, J. Chen, Z. Chen, Z. Weng, Tetrahedron 72 (2016) 3525-3530. |
| [8] |
(a) J.M.R. Narayanam, C.R.J. Stephenson, Chem. Soc. Rev. 40 (2011) 102-113; (b) C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322-5363; (c) J. Xuan, Z.G. Zhang, W.J. Xiao, Angew. Chem. Int. Ed. 54 (2015) 15632-15641; (d) N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075-10166; (e) Q.Q. Zhou, Y.Q. Zou, L.Q. Lu, W.J. Xiao, Angew. Chem. Int. Ed. 58 (2019) 1586-1604; (f) R.S.J. Proctor, R.J. Phipps, Angew. Chem. Int. Ed. 58 (2019) 13666-13699. |
| [9] |
S. Mukherjee, T. Patra, F. Glorius, ACS Catal. 8 (2018) 5842-5846. DOI:10.1021/acscatal.8b01519 |
| [10] |
M. Wang, X. Zhu, X. Zhang, et al., Org. Biomol. Chem. 18 (2020) 5918-5926. DOI:10.1039/D0OB01160F |
| [11] |
F. Scandola, C.A. Bignozzi, M.T. Indelli, Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds, Kluwer Academic, Dordrecht, The Netherlands, 1993.
|
| [12] |
(a) R.F. Renneke, C.L. Hill, J. Am. Chem. Soc. 108 (1986) 3528-3529; (b) R.F. Renneke, C.L. Hill, Angew. Chem. Int. Ed. 27 (1988) 1526-1527; (c) D. Ravelli, S. Protti, M. Fagnoni, Acc. Chem. Res. 49 (2016) 2232-2242; (d) D.M. Schultz, F. Levesque, D.A. DiRocco, et al., Angew. Chem. Int. Ed. 56 (2017) 15274-15278; (e) S.D. Halperin, D. Kwon, M.E. Holmes, et al., Org. Lett. 17 (2015) 5200-5203; (f) J.G. West, D. Huang, E.J. Sorensen, Nat. Commun. 6 (2015) 10093-10099; (g) I.B. Perry, T.F. Brewer, P.J. Sarver, et al., Nature 560 (2018) 70-75; (h) P.J. Sarver, V. Bacauanu, D.M. Schultz, et al., Nat. Chem. 12 (2020) 459-467; (i) S. Esposti, D. Dondi, M. Fagnoni, A. Albini, Angew. Chem. Int. Ed. 46 (2007) 2531-2534; (j) M.D. Tzirakis, M. Orfanopoulos, J. Am. Chem. Soc. 131 (2009) 4063-4069; (k) L. Capaldo, M. Fagnoni, D. Ravelli, Chem. Eur. J. 23 (2017) 6527-6530; (l) M. Meanwell, J. Lehmann, M. Eichenberger, R.E. Martin, R. Britton, Chem. Commun. 54 (2018) 9985-9988; (m) P. Fan, C. Zhang, Y. Lan, et al., Chem. Commun. 55 (2019) 12691-12694; (n) Y. Kuang, H. Cao, H. Tang, et al., Chem. Sci. 11 (2020) 8912-8918; (o) P. Fan, Y. Lan, C. Zhang, C. Wang, J. Am. Chem. Soc. 142 (2020) 2180-2186; (p) P. Fan, C. Zhang, L. Zhang, C. Wang, Org. Lett. 22 (2020) 3875-3878. |
| [13] |
(a) J. Dong, X. Wang, Z. Wang, et al., Chem. Sci. 11 (2020) 1026-1031; (b) J. Dong, F. Yue, X. Wang, et al., Org. Lett. 22 (2020) 8272-8277. |
| [14] |
V. De Waele, O. Poizat, M. Fagnoni, A. Bagno, D. Ravelli, ACS Catal. 6 (2016) 7174-7182. DOI:10.1021/acscatal.6b01984 |
| [15] |
(a) C.M. Jensen, K.B. Lindsay, R.H. Taaning, et al., J. Am. Chem. Soc. 127 (2005) 6544-6545; (b) S.A. Moteki, A. Usui, S. Selvakumar, T. Zhang, K. Maruoka, Angew. Chem. Int. Ed. 53 (2014) 11060-11064. |
| [16] |
K. Larsson, L. Rosenhall, Clin. Exp. Allergy 26 (1996) 18-19. |