 2021, Vol. 32
2021, Vol. 32 


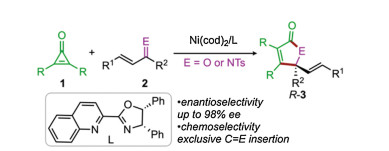
Transition metal-catalyzed cycloaddition reactions have gained tremendous research interest in organic synthesis, which provide one of the most powerful tools for the construction of carbo- and heterocycles [1-4]. In particular, the small ring systems (i.e., cyclopropanes and cyclopropenes) have been widely employed in this type of transformation, due to that the relief of the ring strain would provide the driving force for the C-C oxidative addition to the metal center to afford the key metallacycles [2, 3]. However, despite the significant progress has been witnessed, the development of the enantioselective reactions remains a central challenge in this field. In this context, Li and co-workers very recently reported an elegant example of the nickel-catalyzed [3+2] cycloaddition of α, β-unsaturated ketones/imines with cyclopropenones under relatively mild reaction conditions (Scheme 1) [4]. The salient feature of the reaction is the excellent enantioselectivity and exclusive chemoselectivity. It was found that with the combination of the pre-catalyst Ni(cod)2 and the chiral quinoline-oxazoline ligand L, the reactions of cyclopropenones 1 and α, β-unsaturated ketones/imines 2 can give the [3+2] cycloaddition products R-3 exclusively with excellent enantioselectivity (up to 98% ee), providing a highly efficient strategy for the enantioselective synthesis of the γ-alkenyl butenolides and γ-lactams [5].

|
Download:
|
| Scheme 1. Ni(0)-catalyzed [3+2] cycloaddition of cyclopropenones 1 and α, β-unsaturated ketones/imines 2. | |
{kind=link}
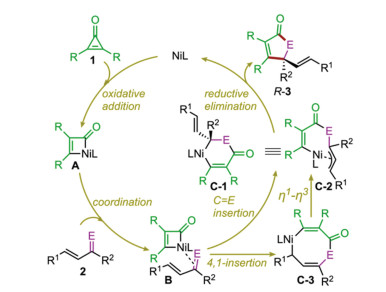
The originally proposed reaction mechanism is depicted in Scheme 2. It was suggested that the reaction is initiated by the C-C oxidative addition of cyclopropenone 1 to give the four-membered nickelacycle A, from which the coordination of the incoming α, β-unsaturated ketone/imine 2 to the Ni center leads to intermediate B. This intermediate can undergo the selective migratory insertion of C=E into the Ni-C(acyl) bond to afford η1-allyl Ni(II) intermediate C-1 or η3-allyl Ni(II) intermediate C-2. The catalytic cycle is then closed by the C-C reductive elimination to forge the [3+2] cycloaddition product. Alternatively, the migratory insertion of 2 into the Ni-C(acyl) bond can occur via a 4, 1-insertion fashion to produce the eight-membered nickelacycle C-3, from which an η1-η3 allylic isomerization can take place to deliver C-2.

|
Download:
|
| Scheme 2. Proposed reaction mechanism. | |
{kind=link}
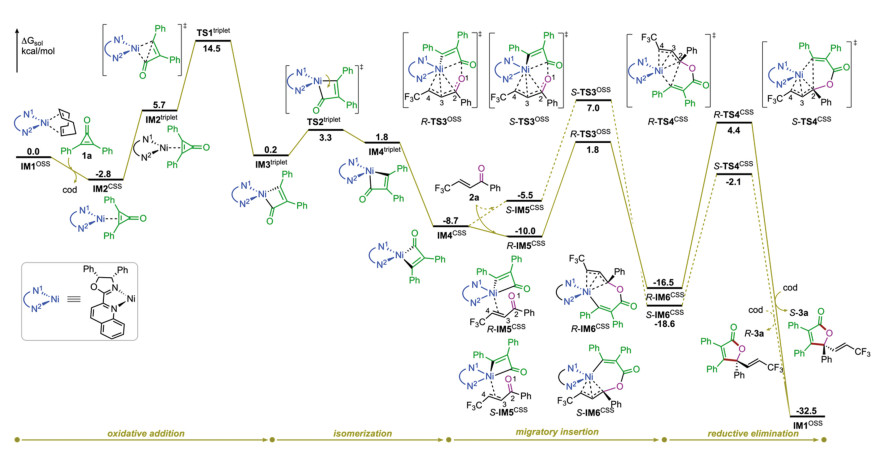
Considering the detailed reaction mechanism and the origins of the enantio- and chemoselectivities remain unclear, we therefore decided to investigate the title reaction computationally by means of density functional theory (DFT) calculations at the level of B3LYP-D3(BJ)-SMD(toluene)/6-311+G(d, p)-SDD//B3LYP-D3(BJ)/6-31G(d)-LANL2DZ (see Supporting information for computational details) [6]. The experimentally used diphenylcyclopropenone 1a and α, β-unsaturated ketone 2a were selected as the model substrates (Fig. 1). To be noted, since that the redox-active quinoline-oxazoline ligand L was employed in the reaction, both singlet (closed- and open-shell) and triplet spin states of all species were considered, and the spin-crossover phenomenon was indeed observed [7]. For the sake of clarity, in the main text we will only present the most favorable pathway, and the other results are provided in Supporting information.

|
Download:
|
| Fig. 1. Calculated energy profile of the Ni(0)-catalyzed [3+2] cycloaddition of diphenylcyclopropenone 1a and α, β-unsaturated ketone 2a. Spin states are indicated in superscript (CSS refers to closed-shell singlet and OSS refers to open-shell singlet). | |
{kind=link}
As shown in Fig. 1, intermediate IM1OSS, which is generated by the highly exergonic ligand exchange process between pre-catalyst Ni(cod)2 and ligand L (see Supporting information for details), was chosen as the starting point of the calculations. The coordination of the C-C double bond of diphenylcyclopropenone 1a to the Ni center of IM1OSS leads to intermediate IM2CSS, which was calculated to be exergonic by 2.8 kcal/mol. The computations show that although IM2triplet is much less stable than IM2CSS (see Supporting information for the minimum energy crossing point), the subsequent C-C oxidative addition was found to occur in the triplet state via transition state TS1triplet rather than that in the singlet state (see Supporting information for details) [8], with an energy barrier of 8.8 kcal/mol relative to IM2triplet (i.e., 17.3 kcal/mol relative to IM2CSS). The C-C oxidative addition was found to be exergonic, and the resulted intermediate four-membered nickelacycle IM3triplet is more stable than IM2triplet by 5.5 kcal/mol.
According to the proposed reaction mechanism (Scheme 2), the next step of the reaction is the migratory insertion of α, β-unsaturated ketone 2a into the Ni-C bond. The reaction occurring directly from IM3triplet was first considered. However, the energy barriers were computed to be relatively high (see the Supporting Information for details). Instead, an isomerization of IM3triplet through the rotation of the Ni-C(alkenyl) bond via transition state TS2triplet was found to take place, requiring an energy barrier of only 3.1 kcal/mol relative to IM3triplet. The resulted nickelacycle IM4triplet, differing in the relative orientation of Ni-C(alkenyl) bond, is slightly higher in energy than IM3triplet by 1.6 kcal/mol.
Though the C-C oxidative addition and the isomerization were computed to occur preferentially at the triplet state, the ensuing reaction steps, i.e., migratory insertion and C-C reductive elimination, were found to take place at the singlet state (see Supporting information for the minimum energy crossing point). Starting from the square planar IM4CSS, which is much more stable than IM4triplet by 10.5 kcal/mol, the possible pathways leading to final [3+2] cycloaddition products R-3a and S-3a were all considered. The computations show that the migratory insertion into the Ni-C(acyl) bond is much more favored than that into the Ni-C(alkenyl) bond (see Supporting information for details). Moreover, the 4, 1-insertion, due to the presence of the additional stabilization of the C3-C4 double bond to the Ni center, was computed to be much more favored than the 2, 1-insertion (i.e., C=O insertion) [9, 10].
The migratory insertion begins with the coordination of the C3-C4 double bond of 2a to the Ni center of IM4CSS. The resulted intermediates R-IM5CSS and S-IM5CSS then undergo the 4, 1-insertion via transition states R-TS3OSS and S-TS3OSS, respectively. Interestingly, the IRC calculations imply that instead of the eight-membered nickelacycles (e.g., C-3, Scheme 2), the 4, 1-insertion leads directly to the η3-allyl Ni(II) intermediates R-IM6CSS and S-IM6CSS, from which the C-C2 reductive elimination can occur readily via transition states R-TS4CSS and S-TS4CSS, delivering the final [3+2] cycloaddition products R-3a and S-3a, respectively. It should be mentioned here that the C-C4 reductive elimination was also considered in the calculations, which could give the experimentally not observed [3+4] cycloaddition products. The results show that the energies of the C-C4 reductive elimination are much higher than those of the C-C2 reductive elimination, thereby ruling out this possibility (see Supporting information for details).
The computations reveal that from intermediate IM4CSS, the C-C reductive elimination is the irreversible step for the R-pathway, while for the S-pathway the irreversible step is the 4, 1-insertion. The enantioselectivity of the reaction is thus determined by the competition between transition states R-TS4CSS and S-TS3OSS. The calculated energy difference of 2.6 kcal/mol (4.4 kcal/mol of R-TS4CSS versus 7.0 kcal/mol of S-TS3OSS) corresponds to a calculated enantioselectivity of 98% ee at the reaction temperature (25 ℃), being in excellent agreement with the experimentally observed 95% ee [4].
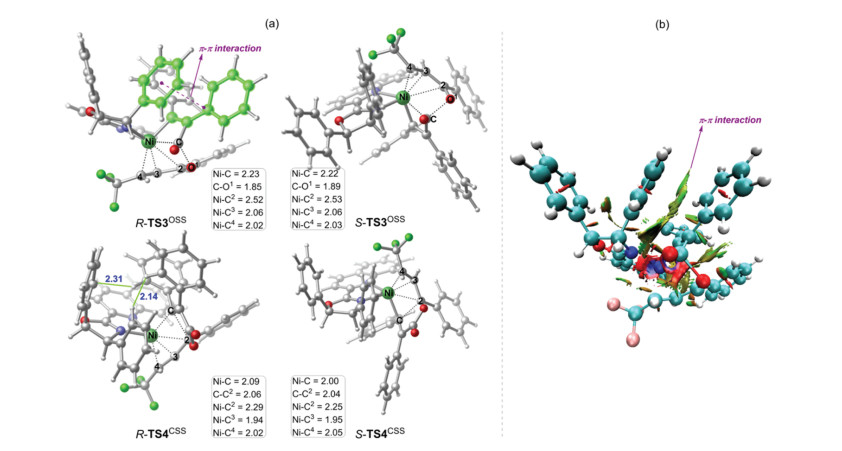
The origins of the enantioselectivity are mainly attributed to the significant preference of 4, 1-insertion transition state R-TS3OSS over S-TS3OSS (1.8 kcal/mol of R-TS3OSS versus 7.0 kcal/mol of S-TS3OSS). The optimized geometries of the key transition states are given in Fig. 2a. The results show that for the 4, 1-insertion, the key bond distances in both R-TS3OSS and S-TS3OSS are quite similar. However, in R-TS3OSS, the diphenylcyclopropenone moiety was found to be rearranged to parallel with the phenyl substituent of the oxazoline ring of the ligand (highlighted in green, Fig. 2). As a consequence, the π-π interaction was observed in R-TS3OSS, as evidenced by the reduced density gradient analysis of R-TS3OSS (Fig. 2b). On the other hand, in S-TS3OSS, no such interaction can be found due to the opposite orientation of the two moieties. Therefore, the presence of the additional π-π interaction makes R-TS3OSS much lower in energy than S-TS3OSS, leading to the experimentally observed enantioselectivity. This argument is also in accordance with the experiments that the absolute configuration of the product was found to be dictated by that of the 5-position in the ligand (i.e., the configuration of the phenyl substituent of the oxazoline ring) [4]. The additional calculations using the ligand with the opposite configuration of the phenyl substituent of the oxazoline ring were further performed to support our argument. The results indeed show that the complementary enantioselectivity should be observed in the reaction (see Supporting information for details).

|
Download:
|
| Fig. 2. (a) Optimized geometric structures of the key transition states. Bond distances are given in Å. (b) Reduced density gradient analysis of R-TS3OSS. | |
{kind=link}
The trend of the reactivity of the C-C2 reductive elimination was found to be opposite to that of the 4, 1-insertion, and R-TS4CSS was calculated to be much higher in energy than S-TS4CSS by 6.5 kcal/mol (4.4 kcal/mol of R-TS4CSS versus -2.1 kcal/mol of S-TS4CSS). The origins of the reactivity difference are mainly due to that in R-TS4CSS, the π-π interaction observed in R-TS3OSS is absent in order to achieve the geometry of the C-C2 reductive elimination. Instead, the steric repulsions between the diphenylcyclopropenone moiety and the phenyl substituents of the oxazoline ring were found (2.14 and 2.31Å, Fig. 2a), thus enabling the opposite reactivity.
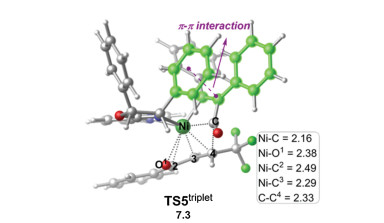
Finally, the chemoselectivity of the C=O versus C=C insertion was investigated. As shown in Fig. 3, the C=C insertion was found to proceed via the 1, 4-insertion through the triplet transition state TS5triplet with an energy barrier of 17.3 kcal/mol relative to R-IM5CSS. The C=C insertion was calculated to be higher in energy than the C=O insertion (via R-TS3OSS) by 5.5 kcal/mol, being in line with the experimentally observed exclusive C=O insertion [4]. Scrutiny of the optimized geometries of R-TS3OSS and TS5triplet (Fig. 2 and 3) shows that the origins of the chemoselectivity could be mainly due to the steric effect. It was found that the distance of the forming C-O1 bond in R-TS3OSS is 1.85Å. However, in TS5triplet, the distance of the forming C-C4 bond is quite long, being 2.33Å, which is probably due to the steric replusion between C4 group and the acyl group is greater than that between the O1 atom and the acyl group. Furthermore, the interactions between the Ni center and the allyl group in R-TS3OSS is stronger than that in TS5triplet (Ni-C2 = 2.52, Ni-C3 = 2.06 and Ni-C4 = 2.02Å in R-TS3OSS versus Ni-C2 = 2.49, Ni-C3 = 2.29 and Ni-O1 = 2.38Å in TS5triplet). The combination of these two factors could result in the energy of R-TS3OSS being much lower than TS5triplet.

|
Download:
|
| Fig. 3. CC insertion via the 1, 4-insertion. Bond distances and energy are given in Å and kcal/mol, respectively. | |
{kind=link}
To summarize, we have herein presented a mechanistic study on the nickel-catalyzed [3+2] cycloaddition of cyclopropenones and α, β-unsaturated ketones by means of DFT calculations. The computations show that the reaction is initiated by the C-C oxidative addition of the cyclopropenone to afford the four-membered nickelacycle, from which an isomerization was required prior to the migratory insertion. The migratory insertion was found to proceed through a 4, 1-insertion fashion to give the η3-allyl Ni(II) intermediate. The catalytic cycle is then completed by the C-C reductive elimination to deliver the [3+2] cycloaddition product. The enantioselectivity of the reaction is determined by the competition between the C-C reductive elimination of the R-pathway and the 4, 1-insertion of the S-pathway. The origins of the enantioselectivity are mainly attributed to the π-π interaction between the diphenylcyclopropenone moiety and the phenyl substituent of the oxazoline ring of the ligand. The experimentally observed exclusive C=O insertion was ascribed to the steric effect. The present results provide a number of new insights into the nickel-catalyzed cycloadditions involving small-ring systems, which should be helpful for the better understanding of related reactions and provide important implications for the future design of new catalytic systems.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (Nos. 22073066, 21503143 and 21975179).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2021.04.006.
| [1] |
(a) R. Peng, Y. Xu, Q. Cao, Chin. Chem. Lett. 29 (2018) 1465-1474; (b) L. Wang, Z. Yu, Chin. J. Org. Chem. 40 (2020) 3536-3558; (c) A. Roglans, A. Pla-Quintana, M. Sola, Chem. Rev. 121 (2021) 1894-1979; (d) J. Wang, S.A. Blaszczyk, X. Li, et al., Chem. Rev. 121 (2021) 110-139. |
| [2] |
(a) P. Chen, B.A. Billett, T. Tsukamoto, et al., ACS Catal. 7 (2017) 1340-1360; (b) R. Vicente, Chem. Rev. 121 (2021) 162-226; (c) M. Murakami, N. Ishida, Chem. Rev. 121 (2021) 264-299. |
| [3] |
(a) M. Murakami, S. Ashida, T. Matsuda, J. Am. Chem. Soc. 127 (2005) 6932-6933; (b) A. Schuster-Haberhauer, R. Gleiter, O. Körner, Organometallics 27 (2008) 1361-1366; (c) P. Chen, T. Xu, G. Dong, Angew. Chem. Int. Ed. 53 (2014) 1674-1678; (d) P. Chen, J. Sieber, C.H. Senanayake, et al., Chem. Sci. 6 (2015) 5440-5445; (e) T. Kondo, Y. Kaneko, Y. Taguchi, et al., J. Am. Chem. Soc. 124 (2002) 6824-6825; (f) T. Lin, C. Zhu, P. Zhang, et al., Angew. Chem. Int. Ed. 55 (2016) 10844-10848; (g) Q. Li, G. Jiang, L. Jiao, et al., Org. Lett. 12 (2010) 1332-1335. |
| [4] |
D. Bai, Y. Yu, H. Guo, et al., Angew. Chem. Int. Ed. 59 (2020) 2740-2744. |
| [5] |
(a) B. Mao, M. Fañanas-Mastral, B.L. Feringa, Chem. Rev. 117 (2017) 10502-10566; (b) S. Wang, Y. Liu, N. Cramer, Angew. Chem. Int. Ed. 58 (2019) 18136-18140; (c) J. Ji, L. Lin, Q. Tang, et al., ACS Catal. 7 (2017) 3763-3767; (d) G.L. Hamilton, E.J. Kang, M. Mba, et al., Science 317 (2007) 496-499. |
| [6] |
(a) Y. Li, Z. Lin, Organometallics 32 (2013) 3003-3011; (b) X. Hong, D. Holte, D.C.G. Götz, et al., J. Org. Chem. 79 (2014) 12177-12184; (c) Y. Liu, Y. Tang, Y. Jiang, et al., ACS Catal. 7 (2017) 1886-1896; (d) S. Yang, Y. Xu, J. Li, Org. Lett. 18 (2016) 6244-6247; (e) Y. Luo, C. Shan, Song Liu, et al., ACS Catal. 9 (2019) 10876-10886; (f) I. Nohira, S. Liu, R. Bai, et al., J. Am. Chem. Soc. 142 (2020) 17306-17311; (g) Y. Li, Y. Luo, L. Peng, et al., Nat. Commun. 11 (2020) 1-13. |
| [7] |
(a) Z. Zhang, J. Zhang, F.K. Sheong, et al., ACS Catal. 10 (2020) 12454-12465; (b) J. Guo, H. Wang, S. Xing, et al., Chem 5 (2019) 1-15; (c) M. Yuan, Z. Song, S.O. Badir, et al., J. Am. Chem. Soc. 142 (2020) 7225-7234; (d) L. Hu, H. Chen, J. Am. Chem. Soc. 139 (2017) 15564-15567; (e) Y. Yu, G. Luo, J. Yang, Catal. Sci. Technol. 9 (2019) 1879-1890; (f) L. Lin, C. Daia, J. Zhu, Org. Chem. Front. 8 (2021) 1531-1543; (g) S. Li, Y. Lan, Chem. Commun. 56 (2020) 6609-6619. |
| [8] |
Y. Luo, C. Shan, S. Liu, ACS Catal. 9 (2019) 10876-10886. |
| [9] |
H. Zou, Z. Wang, G. Huang, Chem. Eur. J. 23 (2017) 12593-12603. |
| [10] |
(a) L. Xu, Q. Zhu, G. Huang, B. Cheng, Y. Xia, J. Org. Chem. 77 (2012) 3017-3024; (b) W. Guo, Y. Xia, J. Org, Chem. 80 (2015) 8113-8121; (c) W. Guo, T. Zhou, Y. Xia, Organometallics 34 (2015) 3012-3020; (d) L. Xu, X. Zhang, M.S. McCammant, et al., J. Org. Chem. 81 (2016) 7604-7611; (e) Y. Liu, Y. Tang, Y.Y. Jiang, et al., ACS Catal. 7 (2017) 1886-1896; (f) G. Lu, R.Y. Liu, Y. Yang, et al., J. Am. Chem. Soc. 139 (2017) 16548-16555; (g) X. Lv, F. Huang, Y.B. Wu, G. Lu, Catal. Sci. Technol. 8 (2018) 2835-2840; (h) X.W. Chen, L. Zhu, Y.Y. Gui, et al., J. Am. Chem. Soc. 141 (2019) 18825-18835; (i) X. Li, X. Ren, H. Wu, et al., Chin. Chem. Lett. 32 (2021) 9-12; (j) H. Zou, Z.L. Wang, Y. Cao, G. Huang, Chin. Chem. Lett. 29 (2018) 1355-1358; (k) X. Li, H. Wu, Z. Wu, G. Huang, J. Org. Chem. 84 (2019) 5514-5523; (l) G. Huang, P. Liu, ACS Catal. 6 (2016) 809-820; (m) M. Zhang, G. Huang, Dalton Trans. 45 (2016) 3552-3557; (n) L. Hu, Z. Wu, G. Huang, Org. Lett. 20 (2018) 5410-5413. |