 2021, Vol. 32
2021, Vol. 32 


b School of Environment, Tsinghua University, Beijing 100084, China;
c Institute of Material Science, University of Connecticut, Storrs 06269, United States
Ozone (O3) is a highly reactive gas consisting of three oxygen atoms. It is both a natural product and a man-made product, and it occurs in the upper atmosphere (stratosphere) and lower atmosphere (troposphere) of the earth. Depending on where it is in the atmosphere, O3 can affect life on Earth in good or bad ways [1, 2]. Stratospheric O3 (approximately 15-30 km above the earth surface) is formed naturally through the interaction of solar ultraviolet (UV) radiation (λ < 210 nm) with molecular oxygen (O2) [3]. The main absorption band of O3 is in the range of 200-300 nm with a maximum wavelength of 254 nm. It reduces the amount of harmful UV radiation reaching the Earth's surface through absorbing most UVB (280-320 nm) and the remaining part of UVC (200-280 nm) that are not blocked by ordinary oxygen in the air [4]. Tropospheric O3 is mainly formed by photochemical reactions between two major types of air pollutants: volatile organic compounds (VOCs) and nitrogen oxides (NOx). These reactions are considered to depend on the presence of heat and sunlight, which lead to higher ambient O3 concentrations during the summer months [5]. For example, an hourly mixing ratio of up to 286 ppb was observed in summer 2005 at a rural mountain site north of Beijing [6]. Long-term exposure to elevated concentrations of tropospheric O3 will cause adverse health effects, including exacerbation of breathing, pulmonary dysfunction, and hospitalization of respiratory diseases [7].
Since people spend almost 90% of their time indoors, the attention to indoor O3 pollution should be reinforced immediately [8]. On one hand, outdoor O3 can enter room directly through ventilation. Indoor-outdoor ratios for O3 depend on both the air exchange rate and the rate at which O3 is removed by indoor surfaces [9]. For example, at 2 air changes per hour, indoor O3 levels are typically 35% of outdoor levels. It exceeds 50 parts per billion (ppb) in places with severe photochemical smog [10]. On the other hand, O3 is also generated by corona discharge. Rooms with printers or electrostatic ventilation system usually emit excess O3. Valuntaite and Girgzdiene [11] reported that the average O3 concentration near the copy machine was 280±63 μg/m3. Moreover, in some specifically confined space, such as cabins [12], hospitals [13] or water treatment plants [14], the concertation of O3 obviously exceeds standard limits (Chinese indoor quality standard, GB/T 18883-2020, below 0.08 ppm, average per 1 h). In addition to the endanger of O3, indoor O3 also reacts rapidly with some organics, especially those containing unsaturated carbon-carbon bonds, which increase the risk of exposure to more toxic pollutants. For example, the concentration formaldehyde, acetaldehyde, and aldehydes involving 5-10 carbons dramatically increase at the presence of O3 [10]. Thus, it is vitally important to study the methods and materials for indoor O3 elimination [15, 16].
Various methods have been attempted to control O3 emission, including physical and chemical adsorption, thermal decomposition, and catalytic decomposition. For example, the modified activated carbon (AC; HNO3 treatment) in Subrahmanyam's work shows the stable O3 conversion as long as 12 h on stream [17]. However, the stability of AC is not long-lasting, since surface carbon will be easily oxidized at the initial stage and the O3 removal efficiency drops dramatically after that. In addition, liquid sodium carbonate or potassium iodide works well for O3 removal [18]. With higher KI loadings (1.2 g), denuders can be used at 20 L/min to remove O3 for several days in the Los Angeles atmosphere under conditions of mild to severe photochemical smog [19]. However, pH condition of the solution needs to be seriously controlled [18], which increases the barrier for its real application. Furthermore, O3 can also be thermally decomposed within 1.5 s at the temperature of 250 ℃ [2]. The high energy consumption and the serious influence of indoor comfort make this technique inapplicable. UV-assisted photocatlysis is another strategy that can eliminate indoor O3 [20, 21]. In addition, it is also effective for indoor bio-aerosols disinfection [22]. However, this technique needs relatively long residence time (seconds level). Considering indoor ventilation efficiency, the O3 removal catalysts used in the ventilation system need to be workable at a very short residence time (a few hundredths of a second). Room-temperature catalysis using metals or metal oxides (e.g., MnO2, NiO, Pd, Ag) seems to be more attractive for the removal of O3 due to its advantages of mild reaction conditions, high efficiency and low energy consumption.
In this review, we put more emphasis on the room-temperature airborne O3 decomposition catalysts, which includes zeolite, metal organic frameworks (MOFs), metal oxides, and noble metals. The active sites and deactivation mechanism are also elaborated. We hope this review will be helpful to guild the modification of current catalysts and highlight prospect in developing new catalysts regarding the long-term O3 elimination, especially in the real environment.
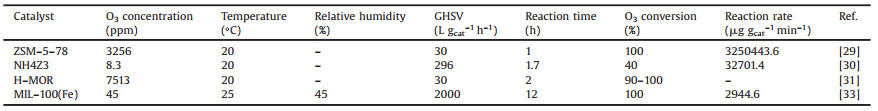
2. Room-temperature catalysisCatalysis includes homogeneous and heterogeneous reaction. Homogeneous O3 decomposition mostly occurs in stratosphere with free radicals, such as hydroxyl radical (·OH), nitric oxide radical (NO·), chlorine radical (Cl·) and bromine radical (Br·) [23, 24]. This part will not be discussed here because the purpose of this review focuses on the control of indoor O3. Heterogeneous O3 catalysis, especially room-temperature catalysts, can be used in the ventilation system or used as indoor cleaner to reduce indoor O3 concentration. Since the 1970s, scholars in various countries have carried out the research of O3 decomposition with heterogeneous catalysts. Li et al. [25, 26] studied the O3 decomposition mechanism over MnO2/γ-Al2O3 catalyst by in-situ Raman and 18O isotope substitution experiment. The O3 decomposition mechanism under dry condition can be simply described as follows (Fig. 1a): First, O3 undergoes dissociative adsorption on the surface to form an oxygen molecule and an atomic oxygen; Then, the atomic oxygen further reacts with the gaseous O3 molecule to form an adsorbed O22- and gaseous oxygen molecules; Finally, the O22- intermediate is decomposed to form oxygen molecules and desorbed from the catalyst surface. However, when the relative humidity is high, the entire mechanism seems to be completely changed (Fig. 1b). Water first dissociates at active site to form surface OH. Then, O3 might react with surface OH with the release of HO3·. Then HO3· will be transformed into ·OH, and ·OOH. Finally, another O3 will react with ·OOH with the generation of H2O and O2. The former mechanism is widely accepted while the latter one is still doubtable and needs further evidence.

|
Download:
|
| Fig. 1. O3 decomposition mechanism under (a) dry and (b) humid condition. | |
Up to now, the active components of the solid catalysts include zeolite, metal organic frameworks (MOFs), metal oxides, noble metals and so on. Most of the reported catalysts can decompose O3 at ambient temperature and exhibit good stability [1, 2, 15, 16]. The category and advantages/disadvantages of different types of catalysts are shown in Table 1. Details of the catalysts will be discussed in the following part.
|
|
Table 1 The category and advantages/disadvantages of different types of room-temperature O3 decomposition catalysts. |

Zeolite with microporous structure can work at room temperature for O3 decomposition. More and more researches ascribe the active site of zeolite to Lewis acid sites, because O3 could act like a Lewis base according to its resonance structure [27]. Lewis acidity in a zeolite generally results from isolated aluminum, which can accept electron pairs due to its uncoordinated structure [28]. As a result, the content of Lewis acid sites in a tetrahedral zeolite framework can be adjusted by using aluminum (Al) substitutes for silicon (Si). Brodu et al. [29] synthesized ZSM-5 zeolites with three ratios of Si/Al (78, 360, 2100). ZSM-5 with lowest Si/Al ratio, i.e., highest acid site, exhibits the best performance for O3 decomposition. The strength of acid site was also investigated. ZSM-5 with lowest Si/Al ratio features 17% strong acid site, which is favorable for O3 decomposition. The strong Lewis acid sites favor the decomposition of O3, whereas the weak acid sites adsorb O3 molecular. In addition, ammonium ion-exchange treatment follows thermal treatment can also adjust the density and strength of Lewis acid sites [30]. Furthermore, the pore size is also very important for the diffusion of O3 to the active site in Zeolite. Brodu et al. [31] studied the effect of zeolite framework and pore width on the efficiency of decomposition of gaseous O3. Compared with microporous zeolite (H-FAU) and interconnected channels zeolite (H-MFI, Na-MFI), parallel channels H-MOR displays the highest efficiency for O3 decomposition. It shows channels or cages with slightly larger size than that of O3 molecules promote the interaction between O3 and Lewis acid sites. Although lots of modifications have been made on zeolite, the O3 decomposition activity of zeolite is still limited due to the intimate occupation of strong Lewis acid sites by peroxide and atomic oxygen.
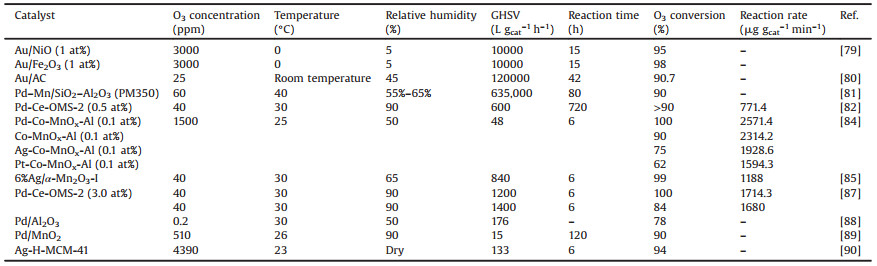
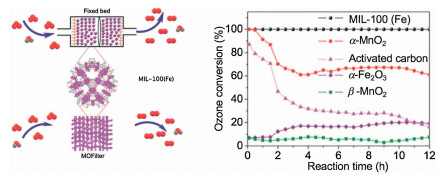
3.2. MOFsMetal-organic frameworks (MOFs) are a class of compounds consisting of metal ions or clusters that coordinate with organic ligands to form one-, two-, or three-dimensional structures [32]. Wang et al. [33] proposed an iron-containing metal organic framework, MIL-100(Fe), for O3 removal. In Fig. 2, the results show that MIL-100(Fe) exhibits 100% persistent O3 conversion efficiency in 100 h at 45% relative humidity and space velocity of 1.9 × 105 h-1 at room temperature. The high efficiency and long-term O3 decomposition activity are ascribed to its porous structure and highly dispersed Fe3+ active sites. More interestingly, presence of water is favorable for O3 elimination, and decomposition rate of O3 is enhanced and with relative humidity ranging from 40% to more than 90%. The O3 decomposition activity over zeolites and MOFs are displayed in Table 2.

|
Download:
|
| Fig. 2. The structure and O3 decomposition activity of MIL-100(Fe). Reproduced with permission [33]. Copyright 2018, Wiley. | |
|
|
Table 2 The structure and O3 decomposition activity of MIL-100(Fe). Reproduced with permission [33]. Copyright 2018, Wiley. |
Metal oxides-type of O3 catalysts have been rapidly developed during the last 5 years. Heisig et al. [34] found the the order of catalytic activity of different materials followed: MnO2 > Co3O4 > NiO > Fe2O3 > Ag2O > Cr2O3 > CeO2 > MgO > V2O5 > CuO > MoO3, when experiments were carried out at a temperature of 313 K, O3 concentration of 2 ppm, relative humidity of 40%, and flow rate of 1.8 L/s. In this part, manganese oxides especially MnO2, presenting outstanding O3 decomposition activity are mainly discussed among all kinds of metal oxides.
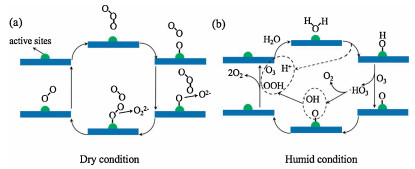
3.3.1. Manganese oxidesJia et al. [35, 36] found that the crystal structure and morphology of manganese dioxide (MnO2) have a significant impact on its catalytic activity for the decomposition of O3. Among them, the activity of α-MnO2 is better than that of β-MnO2 and γ-MnO2, and the activity of nanofiber α-MnO2 is higher than that of nanorod and tubular α-MnO2. Oxygen vacancy (VO) is the decisive factor of catalytic activity. The content and the disperse of VO in MnO2 affect the catalytic activity of O3 decomposition. The function of VO is shown in Fig. 3. Through the in-situ Raman observation, the decomposition of the peroxide species (O22-) on the catalyst surface is the rate-limiting step. Liu et al. [37] also ascribed the higher activity of amorphous MnOx to its abundant VO. In addition, larger specific surface area and mixed Mn2+, Mn3+ and Mn4+ in the amorphous mesoporous MnOx also facilitate the O3 decomposition rate.

|
Download:
|
| Fig. 3. Illustration of VO in O3 decomposition process. Reproduced with permission [35]. Copyright 2016, Elsevier. | |
Since VO in MnO2 is the decisive factor for O3 decomposition, more and more work focus on the generation of VO on MnO2. Zhu et al. [38] obtained α-MnO2 nanofibers with high surface VO by vacuum deoxidation method. The formation of surface VO greatly improves the adsorption and decomposition of O3 molecules on the surface of α-MnO2. After that, Zhu et al. [39] treated α-MnO2 nanowires with adjustable K+ concentration through hydrothermal process in KOH solution. The results indicate that the catalyst life increases from 3 h to 15 h comparing with the untreated sample. This is mainly due to the electrostatic interaction between the oxygen atom in the tunnel and the introduced K+, which results in a large number of active VO, thus improving the performance of the catalyst. Similar phenomena are also observed on Na+ doped OMS-2 sample [40]. The introduction of Na+ in the tunnel framework of OMS-2 facilitates lattice defect formation. In addition, the substitution of K+ by larger or smaller ions such as protons and Ag+ can also result in more VO [41, 42]. For example, Ag+ can be well dispersed in the microtunnels of α-MnO2 without destroying the original tunnel structure. The nonstoichiometric changes in the local structure increase the presences of VO [42]. Furthermore, element doping is a traditional and effective strategy that can optimize electronic and geometric and structures of catalytic centers [43]. Jia et al. [44] prepared iron-modified manganese oxide (Fe-MnOx), and found the obtained Fe-MnOx catalyst contained more VO, which made O3 decomposition rate high in dry/humid air. Li et al. [45] synthesized the metal (cerium and cobalt) doped γ-MnO2 catalyst for O3 decomposition. The results show that the O3 conversion rate follows: Ce-γ-MnO2 (96%) > Co-γ-MnO2 (55%) > γ-MnO2 (38%). The introduction of Ce creates more VO, and also increases the specific surface area, which facilitates O3 adsorption and decomposition. Ma et al. [46] used cryptomelane as a model and further investigated the doping mechanism. Co3+ and Fe3+ replace Mn3+ in the structure of cryptomelane, while Ce4+ mainly replaces K+ in the tunnel and partially replaces Mn4+ in the structure. The former inhibits O3 decomposition while the later facilitates, because the content of Mn3+ and surface VO play key roles in O3 decomposition. Furthermore, the introduction of tinstone (W) and vanadium (V) on α-MnO2 also promote the O3 decomposition activity [47, 48]. MnO, Mn2O3 and MnCO3 also display O3 decomposition activity [49, 50, 51, 52]. For example, Yu et al. [49] fabricated AC based MnO catalyst. The minimum size of MnO nanoparticles and the maximum surface VO synthesized at 700 ℃ of calcination are responsible for the enhanced O3 decomposition rate. Ma et al. [50] found α-Mn2O3 can also decompose O3, and it activity can be increased by a factor of 2.5 with the addition of Ce. The O3 decomposition activity over manganese-based catalysts are shown in Table 3 [35-40, 44-51, 53-68].
|
|
Table 3 The O3 decomposition activity over manganese-based catalysts. |

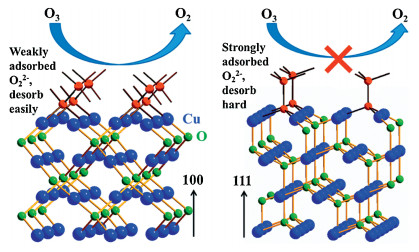
Mathew et al. [69] prepared a mesoporous two-line ferrihydrite (M2LFh), whereas unsaturated FeOx species are widely existed on the surface. M2LFh exhibits about 43.5 times higher O3 removal rate than commercial Fe2O3 at room temperature. The tricoordinated Fe site can result in more deformation of O3 molecular comparing with tracoordinated one (O(Ⅰ)-O(Ⅱ) distance: 1.52 vs. 1.37 Å), which facilitates O3 decomposition. Gong et al. [70] prepared Cu2O nanoparticles with different exposed facets, i.e., (100) and (111) (Fig. 4). Cu2O with (100) plane has the higher O3 conversion efficiency comparing with (111) facet, because O22- adsorbs onto the (100) plane by two shared bonds, which is weaker than the three bonds on (111). NiO can also decompose O3 effectively. However, it activity will be inhibited in the presence of water due to its strong intimation with water, resulting in the block of surface active sites. When Ni is introduced, on one hand, the electron transfer from NiO to Ni may promote the electron release of O22-; one the other hand, lattice expansion in Ni-supported NiO caused by lattice mismatch is good for the movement of adsorbed OH, which can weaken competitive adsorption of H2O [71]. Wang et al. [72] created a lot of VO on ZnO, and revealed the relationship between catalyst activity and surface VO. Modified ZnO by H2 reduction and Ga/Li doping can maintain 30% O3 degradation rate at 6 h for 20 ppm of O3 and 240, 000 mL g-1 h-1 of GHSV.

|
Download:
|
| Fig. 4. Schematics of O3 decomposition on (100) and (111) surface planes of Cu2O. Reproduced with permission [70]. Copyright 2017, Royal Society of Chemistry. | |
The carrier may affect the geometric and electronic structure of the surface-active components. Under the same preparation conditions, MnOx displays monomeric type on Al2O3 supported sample while it shows multinuclear structure for the other supports. In addition, the support also results in different numbers of empty d-states of MnOx, affecting the entire O3 decomposition rate [73]. Carbon material is a commonly used support. Ji et al. [74] immobilized MnOx on carbon nanotubes. The large specific surface area and enhanced ability of electron transfer from CNTs to MnOx might be responsible for the enhanced activity towards O3 decomposition. Similar phenomena were also reported on Graphene based Cu2O hybrid catalyst, easy electron transfer at contacted interface promotes O3 decomposition [75]. Besides, based on the concept of Lewis acid sites and surface VO, O3 tends to be nucleophilic rather than electrophilic. The promoted activity maybe the synergistic effect from both metal oxides and carbon materials.
Using bimetallic oxides for O3 decomposition were also investigated. Heisig et al. [34] loaded the above-mentioned active single metal oxides and bimetallic oxides on the activated carbon, and found that the activity of bimetallic oxides was significantly higher than that of single metal oxides. The order of activity was as follows: Mn-Fe (87%) > Co-Fe (80%) > Mn-CO (78%) > Mn-Ni (74%) > Fe-Ni (71%) > Co-Ni (55%). Because of the interaction between MnO2 and Fe2O3, Mn-Fe system showed the best catalytic activity for O3 decomposition. In addition, perovskite-type mixed oxides, such as LaFeO3 was also prepared and tested O3 decomposition activity. Comparing with commercial Fe2O3, LaFeO3 shows higher O3 decomposition both in dry and humid conditions. It is because LaFeO3 shows no structure deformation, no O22- accumulation, high density of acid site, and easy water desorption. Moreover, the replacement of Fe atom by Ni increases the content of Fe2+, promoting O3 decomposition [76]. The O3 decomposition activity over manganese-free metal oxides mentioned above are shown in Table 4 [69-72, 75-77].
|
|
Table 4 The O3 decomposition activity over manganese-free metal oxides. |

Supported nobel metals are efficient catalysts for the decomposition of gaseous O3 [1, 78]. For example, Hao et al. [79] reported Au/Fe2O3 prepared by precipitation have good O3 removal activity. When the temperature was 0 ℃, the relative humidity was 5%, the initial concentration of O3 was 3000 ppm, and the GHSV was 10, 000 h-1, the catalytic activity of O3 decomposition was as high as 98% after 15 h reaction while single Fe2O3 dropped to 41% within 1 h. Zhang et al. [80] loaded Au particles on coal-based AC (Au/AC) and tested its O3 decomposition activity in a more realistic condition. In the case of high space velocity of 120, 000 h-1, O3 concentration of 50 mg/m3, and relative humidity of 45%, the Au/AC catalyst still exhibits 90.7% O3 removal efficiency as long as 2500 min. Promoted activity is also observed on Pd-based catalyst [81, 82]. However, the high price of the noble metals restricts wide application and has been encouraged the use of less expensive catalysts. Ag catalyst especially in the state of Ag2O, exhibits obviously better activity comparing with commercial Fe2O3, Co3O4, CeO2, Mn2O3, CuO, Pb2O3, Bi2O3, SnO2, MoO3, V2O5 catalyst when the O3 concentration is 3000 ppm; GHSV is 9000 h-1; reaction time is 10 min [83]. The advantage of precious metal catalysts is that they can maintain high O3 decomposition activity under high relative humidity conditions. Tao et al. [84] found Pd-Co-MnOx/Al2O3 catalyst exhibits high moisture tolerance. The introduction of Pd weakens H2O adsorption on the catalyst surface. Similar phenomenon was also observed on Ag modified MnO2 catalysts [85, 86]. The O3 decomposition activity over noble metal catalysts are shown in Table 5 [79-82, 84, 85, 87-90]. The influence of the noble metals on the moisture resistance will be systematically discussed in Part 4.
|
|
Table 5 The O3 decomposition activity over noble metal catalysts. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
From the above analysis, the active sites of O3 decomposition catalysts are described as Lewis acid sites (e.g., zeolite), metal sites (e.g., FeOx, or MOFs) and VO sites (e.g., MnO2) in the previous work. Here, we try to ascribe all the active sites to unsaturated metal sites. For example, the unsaturated coordination structure of Al in zeolite makes it feature extra empty orbit to accept electron pairs from O3. Other researchers also regard unsaturated metal atom, such as Fe atom in FeOx and Fe-MOFs, as active site and use this site to analyze O3 adsorption energy and reaction pathways [33, 46, 76]. For MnO2 catalyst, all literatures classify the O3 active site as VO. Considering that O3 can interact with adjacent manganese atoms after being adsorbed at VO, thus, we regard manganese atoms that are not saturate coordinated as active sites for O3 decomposition. Unsaturated metal sites with different geometric and electronic states usually exhibit various catalytic properties. For example, Li et al. [53] proposed there are two kinds of VO on α-MnO2 (sp2 hybridized VO and sp3 hybridized VO), showing different characteristics for the decomposition of O3. sp2-VO is more intimate to oxygen species. The adsorbed oxygen at sp2 hybridized VO site is difficult to be desorbed, making catalyst deactivated. Thus, it is not accurate to ascribe O3 decomposition active site to all unsaturated metal atoms.
Considering unoccupied d orbitals in metal active sites are Lewis acid sites that can accept electron pairs from O3, we further explain O3 decomposition active sites from the perspective of Lewis acid-base pairs. MnO2 catalyst with VO (removing an O atom will leave behind two unpaired electrons) is regarded as Lewis base. The presence of Lewis base on MnO2 will increase the binding energy with a Lewis acid site (Mn site vacated by the VO), i.e., the strength of Lewis acid will be weakened [91]. Jia et al. also reported that the presence of VO resulted in the weaken of Lewis acid site, demonstrated by NH3-TPD [35]. This phenomenon can also be explained by the increased content of Mn3+ due to VO [35, 38]. The strength of Lewis acid for Mn3+ is 1.698, while it increases to 1.874 for Mn4+, further confirming the VO's negative effect on the strength of Lewis acid [92]. Lewis acid site facilitates to O3 adsorption and decomposition [29-31]. However, too strong Lewis acid site might suppress its activity. Suitable Lewis acid site benefits for O3 polarization and subsequent electron transfer.
4.2. Deactivation mechanismDeactivation is one of the biggest challenges in the use of catalyst. In the process of O3 decomposition, deactivation mainly includes two aspects: accumulation of O22- and inhibition by moisture.
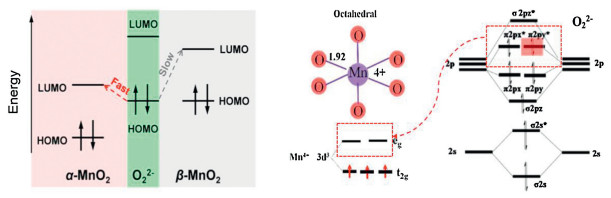
4.2.1. Accumulation of O22-In the dry atmosphere, the rate-limiting step of O3 decomposition on the catalysts is the desorption of adsorbed O22- intermediate to produce O2 [25, 26]. This step requires the electron transfer from the adsorbed species (i.e., O22-) to the active center (i.e., unsaturated metal site). For example, Chen et al. [54] compared two kinds of tunneled MnO2, i.e., α-MnO2 and β-MnO2. α-MnO2 processes higher O3 decomposition activity comparing with β-MnO2. The gap of HOMO (O22-)-LUMO (Mn4+) governs the electron transfer. The eg orbitals of Mn4+ can accept electrons from O22-. The α-MnO2 features the down-shifted 2p3/2 → 3d (eg) transition, which reduces the energy gap between HOMO of O22- and LUMO of Mn4+, i.e., benefiting for electron transfer [93]. The schematic illustration of the relative energy levels of the HOMO and LUMO, and the orbitals of the O22- intermediate and Mn4+ are shown in Fig. 5.

|
Download:
|
| Fig. 5. Schematic illustration of electron transfer from the relative energy levels of the HOMO and LUMO of the O22- HOMO to Mn4+ LUMO. Reproduced with permission [54, 93]. Copyright 2018, ACS & 2018, Elsevier. | |
{kind=link}
The accumulation of O22- can be observed through spectroscopy methodology. High contents of adsorbed O22- and atomic oxygen species are observed ZSM-5 zeolite via in-situ Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), which occupy Lewis acid site and are harmful to the catalyst [29]. In addition, obvious O22- related species were also observed on MnO2 catalyst via in-situ Raman and DRIFTS and the valance state of MnO2 increases as the reaction proceeds, inhibiting the electron transfer from O22- species to Mn3+/Mn4+. Strategies that are commonly used to overcome the accumulation of O22- are as follows:
(a) Modification of fresh catalysts. In order to facilitate electron transfer, the reducibility of the active center needs to be modified. For example, doping MnO2 with Fe, Ce, W, V [44-48] can deduce the average oxidation state in MnO2, benefiting for the electron transfer from O22- species to Mn3+/Mn4+. In addition, unstable active sites are easier to combine with O22-, making the desorption of intermediate products more difficult. Hong et al. [40] found that oxygen vacancy formation energy decreased onced Na+ was introduced in the tunnel framework of OMS-2, which can make accumulated O22- species easily desorbed. Furthermore, composite the catalyst with a material with strong electrical conductivity can promote the electron transfer of O22-. Ji et al. [74] found immobilized MnOx on carbon nanotubes can enhance the O22- electron transfer at contacted interface.
(b) Post-treatment of the catalysts. It is generally considered the deactivation of catalyst is accompanied by the filling out of the oxygen vacancy. High temperature treatment can partially restore catalyst activity. For example, Hong et al. [40] used 425 ℃ calcination under N2 atmosphere to treat used Na-OMS-2 and recovered its activity. The calcination temperature can be further decreased by 75 ℃ in MnOx based Ag catalyst [85].
4.2.2. Inhibition of moistureMoisture is another important factor that affects the activity of the catalyst [55, 56]. Take MnO2 catalyst as an example, the adsorption energy of H2O, O2, and O3 on the (110) surface of VO and VO free α-MnO2 is shown in Fig. 6. The adsorption energies of H2O and O3 reduce from -0.5877 to -0.8746 eV, and -0.4448 to -3.3595 eV due to the formation of surface VO. Though O3 molecule dominates the adsorption process, the competitive adsorption of H2O molecule over VO site cannot be ignored [38]. In addition, under high moisture, the process of gas phase ozone decomposition is similar to that of catalytic ozonation in water because water will be condensed on the surface of catalyst, especially under high relative humidity [94].

|
Download:
|
| Fig. 6. The adsorption energy of H2O, O2, and O3 on the (110) surface of VO and VO free α-MnO2. Reproduced with permission [38]. Copyright 2017, Elsevier. | |
{kind=link}
A free radical-based O3 decomposition mechanism might also exist. O3 can react with surface OH with the generation of HO3·, ·OH, ·O2- etc. The competition between H2O and OH can also lead to the deactivation of catalyst. Up to now, lots of strategies have been applied to alleviate the adsorption of water on O3 active sites:
(a) Reduce the hydrophilic functional groups on the catalyst surface and improve the hydrophobic properties. Hydroxyl groups (OH) are widely existed on metal oxides, which can bond with water molecular via hydrogen bonding, displaying hydrophilic property. This may partly block the active sites. Furthermore, OH may react with O3 with the generation of water and inhibit the O3 decomposition. Liu et al. [57] removed surface hydroxyl groups by treating MnO2 at 300 ℃ to increase catalyst's O3 removal efficiency. In addition, the introduction of Ag in MnOx can also change the catalysts' water resistance characteristic. Deng et al. [41] reported that the addition of Ag significantly decreases the interaction between water vapor and the surface of Ag/MnOx catalysts, showing water tolerance property.
(b) Construct a catalyst with suitable pore structures to reduce water adsorption. Li et al. [95] fabricated a porous cerium-doped manganese oxides via reaction between permanganate, dopamine and Ce3+. The introduction of Ce3+ inhibits the growth of MnOx and gives rise to more macrospores and mesoporous pores. Enlarged pores in Ce-MnOx can alleviate the deactivation caused by moisture condensation (usually occurs at mesoporous pores less than 20 nm) and therefore contribute to the O3 decomposition. Moreover, Cao et al. [96] synthesized higher porosity MnO2-based hybrid aerogel using cellulose nanofibers as the framework. The interconnected macrospores facilitate the passage of water molecules. Furthermore, consumed OH on MnO2 by the coverage of cellulose not only reduce the water adsorption but also avoid the generation of surface-adsorbed H2O via the reaction with O3.
(c) Adjust the active sites to balance the adsorption of ozone and water molecules. Hong et al. [97] modified VO by introducing manganese vacancy (VMn) and appropriate amounts of Li+. The modified structure changes the adsorption energies of O3, H2O, and O2 on MnO2 surface. Theoretical calculation shows that VMn and appropriate amounts of Li+ lower the adsorption energy of O2 and H2O molecules dramatically comparing with the unmodified one, but O3 adsorption is not obviously suppressed. This unique structure shows enhanced water tolerance. In addition, Li et al. [53] also calculated O3, H2O, and O2 adsorption energy at two different VO sites on the MnO2 catalysts, i.e., sp2-VO and sp3-VO. The adsorption energies of O3 and H2O both decrease at sp2-VO site, which might help to improve the water tolerance. However, the adsorption energy of O2 increases at sp2-VO site, inhibiting the desorption of O2. Thus, the entire O3 decomposition process needs to be thoroughly considered especially under humid condition.
(d) Explore water-promoted O3 decomposition catalysts. Almost all the reported catalysts display decreased O3 decomposition activity in the presence of moisture because of inevitable competitive-adsorption at active site. Wang et al. [33] found that water is involved in O3 decomposition reaction using Fe-MIL-100 catalyst. The water molecular deprotonates with the formation of hydroxyl at Fe site. The H atom transfer from Fe-OH to O3 molecule, resulting in the formation of Fe-O species and HO3· radical. This step is the rate-limiting step with the barrier of 25.5 kcal/mol, which is the highest energy barrier among all pathways. In addition, a water involved O3 decomposition mechanism was also proposed according the principles of liquid O3 decomposition [98-100]. Thus, exploring MOF-based catalyst might be a new strategy to overcome the catalyst deactivation caused by moisture.
5. PerspectiveThe aim of this review is to summarize high-efficient O3 decomposition catalysts and investigate their potential application in real indoor ventilation system, especially at high relative humidity and short residence time. In order to evaluate the performance of the catalyst more effectively, the O3 concentration tested is up to tens to thousands of ppm level, which is much higher than that exists in actual indoor environment. Considering the wide use of O3, catalysts summarized here can also be used for residual O3 control in the field of industrial catalytic ozonation. Although plenty of O3 decomposition catalysts have been developed, state-of-the-art catalysts for rapid O3 decomposition with long-term stability under extreme conditions are still not obtained and their corresponding-intrinsic mechanism is still unclear. Future research efforts are suggested as follows:
(a) Active site regulation. MnO2 has been widely studied in O3 catalytic decomposition. At present, it is generally believed that the presence of VO (unsaturated Mn) significantly improves the catalytic activity of MnO2. According to the classic O3 decomposition principle, the desorption of O22- on the surface is a rate-limiting step. Future work should be focused on this part by adjusting the coordination structure to reduce the energy barrier of the rate-limiting step.
(b) The function of water. The presence of water can inhibit the O3 catalytic performance of MnO2. However, Wang et al. [33] found that the water promoted the O3 catalytic activity over Fe-MOF. Based on the mechanism of liquid-phase O3 decomposition, Zhu and Wang et al. [33, 38] proposed a water participated O3 mechanism, but the free radical reactions involved in this mechanism is still a theoretical hypothesis. Sufficient experimental data is still needed.
(c) New catalysts. MOF material is a new type of catalytic material. At present, only Fe-MOF (MIL-100) was investigated in O3 decomposition. The central atom in MOF is the active site. By changing the type of organic ligand, the geometric and electronic structure of center metal varies. In addition, the properties of the ligand itself can regulate the hydrophilic and hydrophobic properties of the MOF, which may be more suitable for its practical applications.
(d) Synergistic effect between O3 and other indoor pollutants. Multiple pollutants coexist in the indoor environment. As an oxidant, O3 can be used to catalytically oxidize indoor VOCs. This strategy can not only solve the indoor VOCs pollution, but also eliminate indoor O3. Furthermore, the oxidation of some VOCs, such as formaldehyde, requires the participation of water, which can alleviate the negative effect of water vapor on most O3 decomposition catalysts.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis project is supported by National Natural Science Foundation of China (Nos. 21707107, 22076150), Fundamental Research Funds for the Central Universities (No. 205208007).
| [1] |
B. Dhandapani, S.T. Oyama, Appl. Catal. B: Environ. 11 (1997) 129-166. DOI:10.1016/S0926-3373(96)00044-6 |
| [2] |
T. Batakliev, V. Georgiev, M. Anachkov, S. Rakovsky, Interdiscip. Toxicol. 7 (2014) 47-59. DOI:10.2478/intox-2014-0008 |
| [3] |
S. Solomon, R.R. Garcia, F.S. Rowland, D.J. Wuebbles, Nature 321 (1986) 755-758. DOI:10.1038/321755a0 |
| [4] |
D. Mitchell, Proc. Natl. Acad. Sci. 103 (2006) 13567-13568. DOI:10.1073/pnas.0605833103 |
| [5] |
M. Shao, Y. Zhang, L. Zeng, et al., J. Environ. Manage. 90 (2009) 512-518. DOI:10.1016/j.jenvman.2007.12.008 |
| [6] |
T. Wang, A. Ding, J. Gao, W.S. Wu, Geophys. Res. Lett. 33 (2006) 1-5. |
| [7] |
M. Jerrett, R.T. Burnett, C.A. Pope Ⅲ, et al., New Engl. J. Med. 360 (2009) 1085-1095. DOI:10.1056/NEJMoa0803894 |
| [8] |
N.E. Klepeis, W.C. Nelson, W.R. Ott, et al., J. Exposure Sci. Environ. Epidemiol. 11 (2001) 231-252. DOI:10.1038/sj.jea.7500165 |
| [9] |
C.J. Cros, G.C. Morrison, J.A. Siegel, R.L. Corsi, Indoor Air 22 (2012) 43-53. DOI:10.1111/j.1600-0668.2011.00734.x |
| [10] |
C.J. Weschler, A.T. Hodgson, J.D. Wooley, Environ. Sci. Technol. 26 (1992) 2371-2377. DOI:10.1021/es00036a006 |
| [11] |
V. Valuntaite, R. Girgždiene, J. Environ. Eng. Landsc. Manage. 15 (2007) 61-67. DOI:10.3846/16486897.2007.9636910 |
| [12] |
C. Weisel, C.J. Weschler, K. Mohan, J. Vallarino, J.D. Spengler, Environ. Sci. Technol. 47 (2013) 4711-4717. DOI:10.1021/es3046795 |
| [13] |
M. Sharma, J.B. Hudson, Am. J. Infect. Control 36 (2008) 559-563. DOI:10.1016/j.ajic.2007.10.021 |
| [14] |
W.H. Glaze, J.W. Kang, D.H. Chapin, Ozone Sci. Eng. 9 (1987) 335-352. DOI:10.1080/01919518708552148 |
| [15] |
X.T. Li, J.Z. Ma, H. He, J. Environ. Sci. 94 (2020) 14-31. DOI:10.1016/j.jes.2020.03.058 |
| [16] |
M. Namdari, C.S. Lee, F. Haghighat, Build. Environ. 187 (2021) 107370. DOI:10.1016/j.buildenv.2020.107370 |
| [17] |
C. Subrahmanyam, D.A. Bulushev, L. Kiwi-Minsker, Appl. Catal. B: Environ. 61 (2005) 98-106. DOI:10.1016/j.apcatb.2005.04.013 |
| [18] |
D. Helmig, Atmos. Environ. 31 (1997) 3635-3651. DOI:10.1016/S1352-2310(97)00144-1 |
| [19] |
E.L. Williams, D. Grosjean, Environ. Sci. Technol. 24 (1990) 811-814. DOI:10.1021/es00076a002 |
| [20] |
J. Kim, P. Zhang, J. Li, J. Wang, P. Fu, Chem. Eng. J. 252 (2014) 337-345. DOI:10.1016/j.cej.2014.05.015 |
| [21] |
J. Patzsch, J.Z. Bloh, Catal. Today 300 (2018) 2-11. DOI:10.1016/j.cattod.2017.07.010 |
| [22] |
S. Josset, J. Taranto, N. Keller, V. Keller, M.C. Lett, Environ. Sci. Technol. 44 (2010) 2605-2611. DOI:10.1021/es902997v |
| [23] |
S. Solomon, Rev. Geophys. 37 (1999) 275-316. DOI:10.1029/1999RG900008 |
| [24] |
B.M. Coldiron, J. Am. Acad. Dermatol. 27 (1992) 653-662. DOI:10.1016/0190-9622(92)70233-6 |
| [25] |
W. Li, G.V. Gibbs, S.T. Oyama, J. Am. Chem. Soc. 120 (1998) 9041-9046. DOI:10.1021/ja981441+ |
| [26] |
W. Li, S.T. Oyama, J. Am. Chem. Soc. 120 (1998) 9047-9052. DOI:10.1021/ja9814422 |
| [27] |
H. Valdes, F.J. Ulloa, V.A. Solar, et al., Microporous Mesoporous Mater. 294 (2020) 109912. DOI:10.1016/j.micromeso.2019.109912 |
| [28] |
P.J. Kunkeler, B.J. Zuurdeeg, J.C. van der Waal, et al., J. Catal. 180 (1998) 234-244. DOI:10.1006/jcat.1998.2273 |
| [29] |
N. Brodu, M.H. Manero, C. Andriantsiferana, J.S. Pic, H. Valdés, Chem. Eng. J. 231 (2013) 281-286. DOI:10.1016/j.cej.2013.07.002 |
| [30] |
H. Valdes, S. Alejandro, C.A. Zaror, J. Hazard. Mater. 227 (2012) 34-40. |
| [31] |
N. Brodu, M.H. Manero, C. Andriantsiferana, J.S. Pic, H. Valdés, Can. J. Chem. Eng. 96 (2018) 1911-1918. DOI:10.1002/cjce.23141 |
| [32] |
J.R. Long, O.M. Yaghi, Chem. Soc. Rev. 38 (2009) 1213-1214. DOI:10.1039/b903811f |
| [33] |
H. Wang, P. Rassu, X. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 16416-16420. DOI:10.1002/anie.201810268 |
| [34] |
C. Heisig, W. Zhang, S.T. Oyama, Appl. Catal. B: Environ. 14 (1997) 117-129. DOI:10.1016/S0926-3373(97)00017-9 |
| [35] |
J. Jia, P. Zhang, L. Chen, Appl. Catal. B: Environ. 189 (2016) 210-218. DOI:10.1016/j.apcatb.2016.02.055 |
| [36] |
J. Jia, P. Zhang, L. Chen, Catal. Sci. Technol. 6 (2016) 5841-5847. DOI:10.1039/C6CY00301J |
| [37] |
S. Liu, J. Ji, Y. Yu, H. Huang, Catal. Sci. Technol. 8 (2018) 4264-4273. DOI:10.1039/C8CY01111G |
| [38] |
G. Zhu, J. Zhu, W. Jiang, et al., Appl. Catal. B: Environ. 209 (2017) 729-737. DOI:10.1016/j.apcatb.2017.02.068 |
| [39] |
G. Zhu, J. Zhu, W. Li, et al., Environ. Sci. Technol. 52 (2018) 8684-8692. DOI:10.1021/acs.est.8b01594 |
| [40] |
W. Hong, T. Zhu, Y. Sun, et al., Environ. Sci. Technol. 53 (2019) 13332-13343. DOI:10.1021/acs.est.9b03689 |
| [41] |
H. Deng, S. Kang, J. Ma, et al., Environ. Sci. Technol. 53 (2019) 10871-10879. DOI:10.1021/acs.est.9b01822 |
| [42] |
T. Gopi, G. Swetha, S. Chandra Shekar, et al., Catal. Commun. 92 (2017) 51-55. DOI:10.1016/j.catcom.2017.01.002 |
| [43] |
L. Miao, J. Wang, P. Zhang, Appl. Surf. Sci. 466 (2019) 441-453. DOI:10.1016/j.apsusc.2018.10.031 |
| [44] |
J. Jia, W. Yang, P. Zhang, J. Zhang, Appl. Catal. A 546 (2017) 79-86. DOI:10.1016/j.apcata.2017.08.013 |
| [45] |
X. Li, J. Ma, L. Yang, et al., Environ. Sci. Technol. 52 (2018) 12685-12696. DOI:10.1021/acs.est.8b04294 |
| [46] |
J. Ma, C. Wang, H. He, Appl. Catal. B: Environ. 201 (2017) 503-510. DOI:10.1016/j.apcatb.2016.08.050 |
| [47] |
Y. Yang, J. Jia, Y. Liu, P. Zhang, Appl. Catal. A 562 (2018) 132-141. DOI:10.1016/j.apcata.2018.06.006 |
| [48] |
Y. Yang, P. Zhang, J. Jia, Appl. Surf. Sci. 484 (2019) 45-53. DOI:10.1016/j.apsusc.2019.04.084 |
| [49] |
Y. Yu, J. Ji, K. Li, et al., Catal. Today (2019) 5090-5099. |
| [50] |
J. Ma, X. Li, C. Zhang, Q. Ma, H. He, Appl. Catal. B: Environ. 264 (2020) 118498. DOI:10.1016/j.apcatb.2019.118498 |
| [51] |
J. Jia, P. Zhang, Ozone Sci. Eng. 40 (2018) 21-28. DOI:10.1080/01919512.2017.1328272 |
| [52] |
X. Wang, Y. Li, Mater. Chem. Phys. 82 (2003) 419-422. DOI:10.1016/S0254-0584(03)00263-3 |
| [53] |
X.T. Li, J.Z. Ma, C.B. Zhang, R.D. Zhang, H. He, J. Environ. Sci. 91 (2020) 43-53. DOI:10.1016/j.jes.2019.12.004 |
| [54] |
Y. Chen, W. Qu, C. Li, et al., Ind. Eng. Chem. Res. 57 (2018) 12590-12594. DOI:10.1021/acs.iecr.8b03491 |
| [55] |
Y. Liu, W. Yang, P. Zhang, J. Zhang, Appl. Surf. Sci. 442 (2018) 640-649. DOI:10.1016/j.apsusc.2018.02.204 |
| [56] |
R. Cao, P. Zhang, Y. Liu, X. Zheng, Appl. Surf. Sci. 495 (2019) 143607. DOI:10.1016/j.apsusc.2019.143607 |
| [57] |
Y. Liu, P. Zhang, J. Phys. Chem. C 121 (2017) 23488-23497. DOI:10.1021/acs.jpcc.7b07931 |
| [58] |
Y. Liu, P. Zhang, Appl. Catal. A 530 (2017) 102-110. DOI:10.1016/j.apcata.2016.11.028 |
| [59] |
Y. Yu, S. Liu, J. Ji, H. Huang, Catal. Sci. Technol. 9 (2019) 5090-5099. DOI:10.1039/C9CY01426H |
| [60] |
Y. Liu, P. Zhang, J. Zhan, L. Liu, Appl. Surf. Sci. 463 (2019) 374-385. DOI:10.1016/j.apsusc.2018.08.226 |
| [61] |
Z. Lian, J. Ma, H. He, Catal. Commun. 59 (2015) 156-160. DOI:10.1016/j.catcom.2014.10.005 |
| [62] |
C. Wang, J. Ma, F. Liu, H. He, R. Zhang, J. Phys. Chem. C 119 (2015) 23119-23126. DOI:10.1021/acs.jpcc.5b08095 |
| [63] |
L. Tao, G. Zhao, P. Chen, et al., ChemCatChem 11 (2019) 1131-1142. DOI:10.1002/cctc.201801401 |
| [64] |
T. Gopi, G. Swetha, S.C. Shekar, et al., Catal. Commun. 92 (2017) 51-55. DOI:10.1016/j.catcom.2017.01.002 |
| [65] |
L. Yang, J. Ma, X. Li, et al., J. Environ. Sci. 87 (2020) 60-70. DOI:10.1016/j.jes.2019.06.007 |
| [66] |
X. Li, J. Ma, C. Zhang, R. Zhang, H. He, J. Environ. Sci. 91 (2020) 43-53. DOI:10.1016/j.jes.2019.12.004 |
| [67] |
L. Yang, J. Ma, X. Li, C. Zhang, H. He, Ind. Eng. Chem. Res. 59 (2020) 118-128. DOI:10.1021/acs.iecr.9b05967 |
| [68] |
L. Yang, J.Z. Ma, X.T. Li, et al., J. Environ. Sci. 87 (2020) 60-70. DOI:10.1016/j.jes.2019.06.007 |
| [69] |
T. Mathew, K. Suzuki, Y. Ikuta, et al., Angew. Chem. 123 (2011) 7519-7522. DOI:10.1002/ange.201102007 |
| [70] |
S. Gong, W. Li, Z. Xie, et al., New J. Chem. 41 (2017) 4828-4834. DOI:10.1039/C7NJ00253J |
| [71] |
S. Gong, A. Wang, Y. Wang, et al., ACS Appl. Nano Mater. 3 (2020) 597-607. DOI:10.1021/acsanm.9b02143 |
| [72] |
A.Q. Wang, L. Zhang, M.G. Rahimi, et al., Appl. Catal. B: Environ. 277 (2020) 119223. DOI:10.1016/j.apcatb.2020.119223 |
| [73] |
R. Radhakrishnan, S.T. Oyama, J.G. Chen, K. Asakura, J. Phys. Chem. B 105 (2001) 4245-4253. DOI:10.1021/jp003246z |
| [74] |
J. Ji, Y. Fang, L. He, H. Huang, Catal. Sci. Technol. 9 (2019) 4036-4046. DOI:10.1039/C9CY00762H |
| [75] |
S. Gong, J. Chen, X. Wu, N. Han, Y. Chen, Catal. Commun. 106 (2018) 25-29. DOI:10.1016/j.catcom.2017.12.003 |
| [76] |
S. Gong, Z. Xie, W. Li, et al., Appl. Catal. B: Environ. 241 (2019) 578-587. DOI:10.1016/j.apcatb.2018.09.041 |
| [77] |
S. Gong, X. Wu, J. Zhang, N. Han, Y. Chen, CrystEngComm 20 (2018) 3096-3104. DOI:10.1039/C8CE00203G |
| [78] |
S.T. Oyama, Catal. Rev. Sci. 42 (2000) 279-322. DOI:10.1081/CR-100100263 |
| [79] |
Z. Hao, D. Cheng, Y. Guo, Y. Liang, Appl. Catal. B: Environ. 33 (2001) 217-222. DOI:10.1016/S0926-3373(01)00172-2 |
| [80] |
P. Zhang, B. Zhang, R. Shi, Front. Env. Sci. Eng. China 3 (2009) 281-288. DOI:10.1007/s11783-009-0032-5 |
| [81] |
Q. Yu, H. Pan, M. Zhao, et al., J. Hazard. Mater. 172 (2009) 631-634. DOI:10.1016/j.jhazmat.2009.07.040 |
| [82] |
L. Yang, J. Ma, X. Li, et al., Catal. Sci. Technol. 10 (2020) 7671-7680. DOI:10.1039/D0CY01298J |
| [83] |
S. Imamura, M. Ikebata, T. Ito, T. Ogita, Ind. Eng. Chem. Res. 30 (1991) 217-221. DOI:10.1021/ie00049a033 |
| [84] |
L.G. Tao, Z.Q. Zhang, P.J. Chen, et al., Appl. Surf. Sci. 481 (2019) 802-810. DOI:10.1016/j.apsusc.2019.03.134 |
| [85] |
X.T. Li, J.Z. Ma, H. He, Environ. Sci. Technol. 54 (2020) 11566-11575. DOI:10.1021/acs.est.0c02510 |
| [86] |
X. Li, J. Ma, C. Zhang, R. Zhang, H. He, J. Environ. Sci. 80 (2019) 159-168. DOI:10.1016/j.jes.2018.12.008 |
| [87] |
P. Nikolov, K. Genov, P. Konova, et al., J. Hazard. Mater. 184 (2010) 16-19. DOI:10.1016/j.jhazmat.2010.07.056 |
| [88] |
M.C. Wu, N.A. Kelly, Appl. Catal. B 18 (1998) 79-91. DOI:10.1016/S0926-3373(98)00027-7 |
| [89] |
T. Kameya, K. Urano, J. Environ. Eng. 128 (2002) 286-292. DOI:10.1061/(ASCE)0733-9372(2002)128:3(286) |
| [90] |
P. Konova, A. Naydenov, P. Nikolov, N. Kumar, J.PorousMater. 25 (2018) 1301-1308. |
| [91] |
H. Metiu, S. Chre'tien, Z. Hu, B. Li, X. Sun, J. Phys. Chem. C 116 (2012) 10439-10450. DOI:10.1021/jp301341t |
| [92] |
Y. Zhang, Inorg. Chem. 21 (1982) 3889-3893. DOI:10.1021/ic00141a006 |
| [93] |
W. Yang, Y. Zhu, F. You, et al., Appl. Catal. B: Environ. 233 (2018) 184-193. DOI:10.1016/j.apcatb.2018.03.107 |
| [94] |
J. Wang, Y. Dang, A.G. Meguerdichian, et al., Environ. Sci. Technol. Lett. 7 (2020) 48-53. DOI:10.1021/acs.estlett.9b00713 |
| [95] |
L.X. Li, P.Y. Zhang, R.R. Cao, Catal. Sci. Technol. 10 (2020) 2254-2267. DOI:10.1039/D0CY00196A |
| [96] |
R. Cao, L. Li, P. Zhang, J. Harzard. Mater. 407 (2021) 124793. DOI:10.1016/j.jhazmat.2020.124793 |
| [97] |
W. Hong, M.P. Shao, T.L. Zhu, et al., Appl. Catal. B: Environ. 274 (2020) 119088. DOI:10.1016/j.apcatb.2020.119088 |
| [98] |
L. Zhao, J. Ma, Z. Sun, H. Liu, Appl. Catal. B: Environ. 89 (2009) 326-334. DOI:10.1016/j.apcatb.2008.12.009 |
| [99] |
J. Nawrocki, B. Kasprzyk-Hordern, Appl. Catal. B: Environ. 99 (2010) 27-42. DOI:10.1016/j.apcatb.2010.06.033 |
| [100] |
S. Afzal, X. Quan, J. Zhang, Appl. Catal. B: Environ. 206 (2017) 692-703. DOI:10.1016/j.apcatb.2017.01.072 |